
Vapour pressure or equilibrium vapor pressure is the pressure exerted by a vapor in thermodynamic equilibrium with its condensed phases at a given temperature in a closed system. The equilibrium vapor pressure is an indication of a liquid's thermodynamic tendency to evaporate. It relates to the balance of particles escaping from the liquid in equilibrium with those in a coexisting vapor phase. A substance with a high vapor pressure at normal temperatures is often referred to as volatile. The pressure exhibited by vapor present above a liquid surface is known as vapor pressure. As the temperature of a liquid increases, the attractive interactions between liquid molecules become less significant in comparison to the entropy of those molecules in the gas phase, increasing the vapor pressure. Thus, liquids with strong intermolecular interactions are likely to have smaller vapor pressures, with the reverse true for weaker interactions.
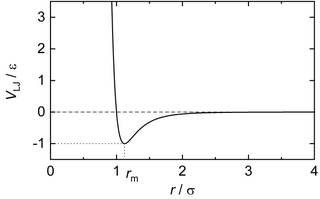
In computational chemistry, the Lennard-Jones potential is an intermolecular pair potential. Out of all the intermolecular potentials, the Lennard-Jones potential is probably the one that has been the most extensively studied. It is considered an archetype model for simple yet realistic intermolecular interactions.
The Dortmund Data Bank is a factual data bank for thermodynamic and thermophysical data. Its main usage is the data supply for process simulation where experimental data are the basis for the design, analysis, synthesis, and optimization of chemical processes. The DDB is used for fitting parameters for thermodynamic models like NRTL or UNIQUAC and for many different equations describing pure component properties, e.g., the Antoine equation for vapor pressures. The DDB is also used for the development and revision of predictive methods like UNIFAC and PSRK.
Jürgen Gmehling is a retired German professor of technical and industrial chemistry at the Carl von Ossietzky University of Oldenburg.

In statistical thermodynamics, the UNIFAC method is a semi-empirical system for the prediction of non-electrolyte activity in non-ideal mixtures. UNIFAC uses the functional groups present on the molecules that make up the liquid mixture to calculate activity coefficients. By using interactions for each of the functional groups present on the molecules, as well as some binary interaction coefficients, the activity of each of the solutions can be calculated. This information can be used to obtain information on liquid equilibria, which is useful in many thermodynamic calculations, such as chemical reactor design, and distillation calculations.

The non-random two-liquid model is an activity coefficient model that correlates the activity coefficients of a compound with its mole fractions in the liquid phase concerned. It is frequently applied in the field of chemical engineering to calculate phase equilibria. The concept of NRTL is based on the hypothesis of Wilson that the local concentration around a molecule is different from the bulk concentration. This difference is due to a difference between the interaction energy of the central molecule with the molecules of its own kind and that with the molecules of the other kind . The energy difference also introduces a non-randomness at the local molecular level. The NRTL model belongs to the so-called local-composition models. Other models of this type are the Wilson model, the UNIQUAC model, and the group contribution model UNIFAC. These local-composition models are not thermodynamically consistent for a one-fluid model for a real mixture due to the assumption that the local composition around molecule i is independent of the local composition around molecule j. This assumption is not true, as was shown by Flemr in 1976. However, they are consistent if a hypothetical two-liquid model is used.
The Joback method predicts eleven important and commonly used pure component thermodynamic properties from molecular structure only.

In statistical thermodynamics, UNIQUAC is an activity coefficient model used in description of phase equilibria. The model is a so-called lattice model and has been derived from a first order approximation of interacting molecule surfaces. The model is, however, not fully thermodynamically consistent due to its two-liquid mixture approach. In this approach the local concentration around one central molecule is assumed to be independent from the local composition around another type of molecule.
A group-contribution method in chemistry is a technique to estimate and predict thermodynamic and other properties from molecular structures.
PSRK is an estimation method for the calculation of phase equilibria of mixtures of chemical components. The original goal for the development of this method was to enable the estimation of properties of mixtures containing supercritical components. This class of substances cannot be predicted with established models, for example UNIFAC.
Process simulation is used for the design, development, analysis, and optimization of technical processes such as: chemical plants, chemical processes, environmental systems, power stations, complex manufacturing operations, biological processes, and similar technical functions.
The Margules activity model is a simple thermodynamic model for the excess Gibbs free energy of a liquid mixture introduced in 1895 by Max Margules. After Lewis had introduced the concept of the activity coefficient, the model could be used to derive an expression for the activity coefficients of a compound i in a liquid, a measure for the deviation from ideal solubility, also known as Raoult's law.
MOSCED is a thermodynamic model for the estimation of limiting activity coefficients. From a historical point of view MOSCED can be regarded as an improved modification of the Hansen method and the Hildebrand solubility model by adding higher interaction term such as polarity, induction and separation of hydrogen bonding terms. This allows the prediction of polar and associative compounds, which most solubility parameter models have been found to do poorly. In addition to making quantitative prediction, MOSCED can be used to understand fundamental molecular level interaction for intuitive solvent selection and formulation.
The Van Laar equation is a thermodynamic activity model, which was developed by Johannes van Laar in 1910-1913, to describe phase equilibria of liquid mixtures. The equation was derived from the Van der Waals equation. The original van der Waals parameters didn't give good description of vapor-liquid equilibria of phases, which forced the user to fit the parameters to experimental results. Because of this, the model lost the connection to molecular properties, and therefore it has to be regarded as an empirical model to correlate experimental results.

DWSIM is an open-source CAPE-OPEN compliant chemical process simulator for Windows, Linux and macOS. DWSIM is built on top of the Microsoft .NET and Mono Platforms and features a Graphical User Interface (GUI), advanced thermodynamics calculations, reactions support and petroleum characterization / hypothetical component generation tools.
Grant McDonald Wilson was a notable American thermodynamicist. He is widely known to the fields of chemical engineering and physical chemistry for having developed the Wilson equation, one of the first attempts of practical importance to model nonideal behavior in liquid mixtures as observed in practice with common polar compounds such as alcohols, amines, etc. The equation has been in use in all commercial chemical process simulators to predict phase behavior and produce safe process designs of commercial and environmental protection importance to the chemical industry. He founded the company Wilco in 1977 to research, measure, commercialize, and publish thermophysical property data for numerous chemical mixtures of interest to the industry. The Journal of Chemical & Engineering Data published a posthumous issue in honor of Wilson in April 2014 in recognition of his extensive contributions to the field.
VTPR is an estimation method for the calculation of phase equilibria of mixtures of chemical components. The original goal for the development of this method was to enable the estimation of properties of mixtures which contain supercritical components. These class of substances couldn't be predicted with established models like UNIFAC.
COSMO-RS is a quantum chemistry based equilibrium thermodynamics method with the purpose of predicting chemical potentials µ in liquids. It processes the screening charge density σ on the surface of molecules to calculate the chemical potential µ of each species in solution. Perhaps in dilute solution a constant potential must be considered. As an initial step a quantum chemical COSMO calculation for all molecules is performed and the results are stored in a database. In a separate step COSMO-RS uses the stored COSMO results to calculate the chemical potential of the molecules in a liquid solvent or mixture. The resulting chemical potentials are the basis for other thermodynamic equilibrium properties such as activity coefficients, solubility, partition coefficients, vapor pressure and free energy of solvation. The method was developed to provide a general prediction method with no need for system specific adjustment.
Computational thermodynamics is the use of computers to simulate thermodynamic problems specific to materials science, particularly used in the construction of phase diagrams.
Thermodynamic modelling is a set of different strategies that are used by engineers and scientists to develop models capable of evaluating different thermodynamic properties of a system. At each thermodynamic equilibrium state of a system, the thermodynamic properties of the system are specified. Generally, thermodynamic models are mathematical relations that relate different state properties to each other in order to eliminate the need of measuring all the properties of the system in different states.
 Latest published parameters
Latest published parameters UNIFAC consortium parameters September 2008
UNIFAC consortium parameters September 2008 In the UNIFAC consortium added or modified parameters
In the UNIFAC consortium added or modified parameters

