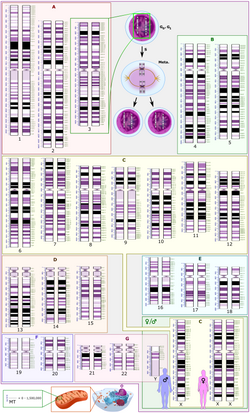
A karyotype (Fig 1) is the characteristic chromosome complement of a eukaryotespecies.[3][4] A karyotype is typically presented as an image of the chromosomes from a single cell arranged from largest (chromosome 1) to smallest (chromosome 22), with the sex chromosomes (X and Y) shown last. Historically, karyotypes have been obtained by staining cells after they have been chemically arrested during cell division. Karyotypes have been used for several decades to identify chromosomal abnormalities in both germline and cancer cells. Conventional karyotypes can assess the entire genome for changes in chromosome structure and number, but the resolution is relatively coarse, with a detection limit of 5-10Mb.[citation needed]
Fig 1. Karyotype of human male using Giemsa staining
Method
Recently, platforms for generating high-resolution karyotypes in silico from disrupted DNA have emerged, such as array comparative genomic hybridization (arrayCGH) and SNP arrays. Conceptually, the arrays are composed of hundreds to millions of probes which are complementary to a region of interest in the genome. The disrupted DNA from the test sample is fragmented, labeled, and hybridized to the array. The hybridization signal intensities for each probe are used by specialized software to generate a log2ratio of test/normal for each probe on the array. [citation needed]
Knowing the address of each probe on the array and the address of each probe in the genome, the software lines up the probes in chromosomal order and reconstructs the genome in silico (Fig 2 and 3).
Virtual karyotypes have dramatically higher resolution than conventional cytogenetics. The actual resolution will depend on the density of probes on the array. Currently, the Affymetrix SNP6.0 is the highest density commercially available array for virtual karyotyping applications. It contains 1.8 million polymorphic and non-polymorphic markers for a practical resolution of 10-20kb—about the size of a gene. This is approximately 1000-fold greater resolution than karyotypes obtained from conventional cytogenetics.[citation needed]
Virtual karyotypes can be performed on germline samples for constitutional disorders,[5][6] and clinical testing is available from dozens of CLIA certified laboratories (genetests.org). Virtual karyotyping can also be done on fresh or formalin-fixed paraffin-embedded tumors.[7][8][9] CLIA-certified laboratories offering testing on tumors include Creighton Medical LaboratoriesArchived 11 February 2010 at the Wayback Machine (fresh and paraffin embedded tumor samples) and CombiMatrix Molecular Diagnostics (fresh tumor samples).
Fig 2. Virtual karyotype of a chronic lymphocytic leukemia sample using a SNP array.Fig 3. Virtual karyotype log2ratio plot of a chronic lymphocytic leukemia sample using a SNP array. Yellow = copy number of 2 (normal/diploid), aqua = 1 (deletion), pink = 3 (trisomy).
Different platforms for virtual karyotyping
Array-based karyotyping can be done with several different platforms, both laboratory-developed and commercial. The arrays themselves can be genome-wide (probes distributed over the entire genome) or targeted (probes for genomic regions known to be involved in a specific disease) or a combination of both. Further, arrays used for karyotyping may use non-polymorphic probes, polymorphic probes (i.e., SNP-containing), or a combination of both. Non-polymorphic probes can provide only copy number information, while SNP arrays can provide both copy number and loss-of-heterozygosity (LOH) status in one assay. The probe types used for non-polymorphic arrays include cDNA, BAC clones (e.g., BlueGnomeArchived 7 February 2014 at the Wayback Machine ), and oligonucleotides (e.g., Agilent, Santa Clara, CA, USA or Nimblegen, Madison, WI, USA). Commercially available oligonucleotide SNP arrays can be solid phase (Affymetrix, Santa Clara, CA, USA) or bead-based (Illumina, San Diego, CA, USA). Despite the diversity of platforms, ultimately they all use genomic DNA from disrupted cells to recreate a high resolution karyotype in silico. The end product does not yet have a consistent name, and has been called virtual karyotyping,[8][10] digital karyotyping,[11] molecular allelokaryotyping,[12] and molecular karyotyping.[13] Other terms used to describe the arrays used for karyotyping include SOMA (SNP oligonucleotide microarrays)[14] and CMA (chromosome microarray).[15][16] Some consider all platforms to be a type of array comparative genomic hybridization (arrayCGH), while others reserve that term for two-dye methods, and still others segregate SNP arrays because they generate more and different information than two-dye arrayCGH methods.[citation needed]
Applications
Detecting copy-number changes
Copy number changes can be seen in both germline and tumor samples. Copy number changes can be detected by arrays with non-polymorphic probes, such as arrayCGH, and by SNP-based arrays. Human beings are diploid, so a normal copy number is always two for the non-sex chromosomes.[citation needed]
Deletions: A deletion is the loss of genetic material. The deletion can be heterozygous (copy number of 1) or homozygous (copy number of 0, nullisomy). Microdeletion syndromes are examples of constitutional disorders due to small deletions in germline DNA. Deletions in tumor cells may represent the inactivation of a tumor suppressor gene, and may have diagnostic, prognostic, or therapeutic implications.
Gains: A copy number gain represents the gain of genetic material. If the gain is of just one additional copy of a segment of DNA, it may be called a duplication (Fig 4). If there is one extra copy of an entire chromosome, it may be called a trisomy. Copy number gains in germline samples may be disease-associated or may be a benign copy number variant. When seen in tumor cells, they may have diagnostic, prognostic, or therapeutic implications.
Fig 4. Schematic of a region of a chromosome before and after a duplication event
Amplifications: Technically, an amplification is a type of copy number gain in which there is a copy number >10. In the context of cancer biology, amplifications are often seen in oncogenes. This could indicate a worse prognosis, help categorize the tumor, or indicate drug eligibility. An example of drug eligibility is Her2Neu amplification and Herceptin, and an image of Her2Neu amplification detected by SNP array virtual karyotyping is provided (Fig 5).Fig 5. Her2 Amplification by SNP array virtual karyotype.
Loss of heterozygosity (LOH), autozygous segments, and uniparental disomy
Autozygous segments and uniparental disomy (UPD) are diploid/'copy neutral' genetic findings and therefore are only detectable by SNP-based arrays. Both autozygous segments and UPD will show loss of heterozygosity (LOH) with a copy number of two by SNP array karyotyping. The term Runs of Homozgygosity (ROH), is a generic term that can be used for either autozygous segments or UPD.[citation needed]
Autozygous segment: An autozygous segment is bi-parental and seen only in the germline. They are extended runs of homozygous markers in the genome, and they occur when an identical haplotype block is inherited from both parents. They are also called "identical by descent" (IBD) segments, and they can be used for homozygosity mapping.[17][18]
Uniparental Disomy: UPD occurs when both copies of a gene or genomic region are inherited from the same parent. This is uniparental, in contrast to autozygous segments which are bi-parental. When present in the germline, they can be harmless or associated with disease, such as Prader-Willi or Angelman syndromes. Also in contrast to autozygosity, UPD can develop in tumor cells, and this is referred to as acquired UPD or copy neutral LOH in the literature (Fig 6). Fig 6. Copy neutral LOH/uniparental disomyAcquired UPD is quite common in both hematologic and solid tumors, and is reported to constitute 20 to 80% of the LOH seen in human tumors.[19][20][21][22] Acquired UPD can serve as the 2nd hit in the Knudson Two Hit Hypothesis of Tumorigenesis, and thus can be the biological equivalent of a deletion.[23] Because this type of lesion cannot be detected by arrayCGH, FISH, or conventional cytogenetics, SNP-based arrays are preferred for virtual karyotyping of tumors.
Fig 7. Virtual karyotype of a colorectal carcinoma (whole genome view) demonstrating deletions, gains, amplifications, and acquired UPD (copy neutral LOH).
Figure 7 is a SNP array virtual karyotype from a colorectal carcinoma demonstrating deletions, gains, amplifications, and acquired UPD (copy neutral LOH).
Examples of clinical cancer applications
A virtual karyotype can be generated from nearly any tumor, but the clinical meaning of the genomic aberrations identified are different for each tumor type. Clinical utility varies and appropriateness is best determined by an oncologist or pathologist in consultation with the laboratory director of the lab performing the virtual karyotype. Below are examples of types of cancers where the clinical implications of specific genomic aberrations are well established. This list is representative, not exhaustive. The web site for the Cytogenetics Laboratory at Wisconsin State Laboratory of Hygiene has additional examples of clinically relevant genetic changes that are readily detectable by virtual karyotyping.Archived 12 April 2009 at the Wayback Machine
Neuroblastoma
Based on a series of 493 neuroblastoma samples, it has been reported that overall genomic pattern, as tested by array-based karyotyping, is a predictor of outcome in neuroblastoma:[24]
Tumors presenting exclusively with whole chromosome copy number changes were associated with excellent survival.
Tumors presenting with any kind of segmental chromosome copy number changes were associated with a high risk of relapse.
Within tumors showing segmental alterations, additional independent predictors of decreased overall survival were MYCN amplification, 1p and 11q deletions, and 1q gain.
Earlier publications categorized neuroblastomas into three major subtypes based on cytogenetic profiles:[25]
Subtype 1: favorable neuroblastoma with near triploidy and a predominance of numerical gains and losses, mostly representing non-metastatic NB stages 1, 2 and 4S.
Subtypes 2A and 2B: found in unfavorable widespread neuroblastoma, stages 3 and 4, with 11q loss and 17q gain without MYCN amplification (subtype 2A) or with MYCN amplification often together with 1p deletions and 17q gain (subtype 2B).
Wilms' tumor
Tumor-specific loss-of-heterozygosity (LOH) for chromosomes 1p and 16q identifies a subset of Wilms' tumor patients who have a significantly increased risk of relapse and death. LOH for these chromosomal regions can now be used as an independent prognostic factor together with disease stage to target intensity of treatment to risk of treatment failure.[26][27]
Chromophobe carcinoma: hypodiploid with loss of chromosomes 1, 2, 6, 10, 13, 17, 21
Array-based karyotyping can be used to identify characteristic chromosomal aberrations in renal tumors with challenging morphology.[8][10] Array-based karyotyping performs well on paraffin embedded tumors[29] and is amenable to routine clinical use.
In addition, recent literature indicates that certain chromosomal aberrations are associated with outcome in specific subtypes of renal epithelial tumors.[30] Clear cell renal carcinoma: del 9p and del 14q are poor prognostic indicators.[31][32] Papillary renal cell carcinoma: duplication of 1q marks fatal progression.[33]
Array-based karyotyping is a cost-effective alternative to FISH for detecting chromosomal abnormalities in chronic lymphocytic leukemia (CLL). Several clinical validation studies have shown >95% concordance with the standard CLL FISH panel.[12][34][35][36][37] In addition, many studies using array-based karyotyping have identified 'atypical deletions' missed by the standard FISH probes and acquired uniparental disomy at key loci for prognostic risk in CLL.[38][39]
Four main genetic aberrations are recognized in CLL cells that have a major impact on disease behavior.[40]
Deletions of part of the short arm of chromosome 17 (del 17p) which target p53 are particularly deleterious. Patients with this abnormality have significantly short interval before they require therapy and a shorter survival. This abnormality is found in 5–10% of patients with CLL.
Deletions of the long arm on chromosome 11 (del 11q) are also unfavorable although not to the degree seen with del 17p. The abnormality targets the ATM gene and occurs infrequently in CLL (5–10%).
Trisomy 12, an additional chromosome 12, is a relatively frequent finding occurring in 20–25% of patients and imparts an intermediate prognosis.
Deletion of 13q14 (del 13q14) is the most common abnormality in CLL with roughly 50% of patients with cells containing this defect. When del 13q14 is seen in isolation, patients have the best prognosis and most will live many years, even decades, without the need for therapy.
Multiple myeloma
Avet-Loiseau, et al. in Journal of Clinical Oncology, used SNP array karyotyping of 192 multiple myeloma (MM) samples to identify genetic lesions associated with prognosis, which were then validated in a separate cohort (n = 273).[41] In MM, lack of a proliferative clone makes conventional cytogenetics informative in only ~30% of cases. FISH panels are useful in MM, but standard panels would not detect several key genetic abnormalities reported in this study.[citation needed]
Virtual karyotyping identified chromosomal abnormalities in 98% of MM cases
del(12p13.31)is an independent adverse marker
amp(5q31.1) is a favorable marker
The prognostic impact of amp(5q31.1) over-rides that of hyperdiploidy and also identifies patients who greatly benefit from high-dose therapy.
Array-based karyotyping cannot detect balanced translocations, such as t(4;14) seen in ~15% of MM. Therefore, FISH for this translocation should also be performed if using SNP arrays to detect genome-wide copy number alterations of prognostic significance in MM.[citation needed]
Medulloblastoma
Array-based karyotyping of 260 medulloblastomas by Pfister S, et al. resulted in the following clinical subgroups based on cytogenetic profiles:[42]
Poor prognosis: gain of 6q or amplification of MYC or MYCN
Intermediate: gain of 17q or an i(17q) without gain of 6q or amplification of MYC or MYCN
Excellent prognosis: 6q and 17q balanced or 6q deletion
Oligodendroglioma
The 1p/19q co-deletion is considered a "genetic signature" of oligodendroglioma. Allelic losses on 1p and 19q, either separately or combined, are more common in classic oligodendrogliomas than in either astrocytomas or oligoastrocytomas.[43] In one study, classic oligodendrogliomas showed 1p loss in 35 of 42 (83%) cases, 19q loss in 28 of 39 (72%), and these were combined in 27 of 39 (69%) cases; there was no significant difference in 1p/19q loss of heterozygosity status between low-grade and anaplastic oligodendrogliomas.[43] 1p/19q co-deletion has been correlated with both chemosensitivity and improved prognosis in oligodendrogliomas.[44][45] Most larger cancer treatment centers routinely check for the deletion of 1p/19q as part of the pathology report for oligodendrogliomas. The status of the 1p/19q loci can be detected by FISH or virtual karyotyping. Virtual karyotyping has the advantage of assessing the entire genome in one assay, as well as the 1p/19q loci. This allows assessment of other key loci in glial tumors, such as EGFR and TP53 copy number status.[citation needed]
Whereas the prognostic relevance of 1p and 19q deletions is well established for anaplastic oligodendrogliomas and mixed oligoastrocytomas, the prognostic relevance of the deletions for low-grade gliomas is more controversial. In terms of low-grade gliomas, a recent study also suggests that 1p/19q co-deletion may be associated with a (1;19)(q10;p10) translocation which, like the combined 1p/19q deletion, is associated with superior overall survival and progression-free survival in low-grade glioma patients.[46] Oligodendrogliomas show only rarely mutations in the p53 gene, which is in contrast to other gliomas.[47]Epidermal growth factor receptor amplification and whole 1p/19q codeletion are mutually exclusive and predictive of completely different outcomes, with EGFR amplification predicting poor prognosis.[48]
Glioblastoma
Yin et al.[49] studied 55 glioblastoma and 6 GBM cell lines using SNP array karyotyping. Acquired UPD was identified at 17p in 13/61 cases. A significantly shortened survival time was found in patients with 13q14 (RB) deletion or 17p13.1 (p53) deletion/acquired UPD. Taken together, these results suggest that this technique is a rapid, robust, and inexpensive method to profile genome-wide abnormalities in GBM. Because SNP array karyotyping can be performed on paraffin embedded tumors, it is an attractive option when tumor cells fail to grow in culture for metaphase cytogenetics or when the desire for karyotyping arises after the specimen has been formalin fixed.[citation needed]
The importance of detecting acquired UPD (copy neutral LOH) in glioblastoma:[citation needed]
Of patients with 17p abnormality, ~50% were deletions and ~50% were aUPD
Both 17p del and 17p UPD were associated with worse outcome.
9/13 had homozygous TP53 mutations underlying the 17p UPD.
In addition, in cases with uncertain grade by morphology, genomic profiling can assist in diagnosis.
Concomitant gain of 7 and loss of 10 is essentially pathognomonic for GBM[50]
EGFR amplification, loss of PTEN (on 10q), and loss of p16 (on 9p) occur almost exclusively in glioblastoma and can provide means to distinguish anaplastic astrocytoma from glioblastoma.[51]
Acute lymphoblastic leukemia
Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, has been increasingly recognized as an important predictor of outcome in acute lymphoblastic leukemia (ALL).[52] NB: Balanced translocations cannot be detected by array-based karyotyping (see Limitations below).
Some cytogenetic subtypes have a worse prognosis than others. These include:
A translocation between chromosomes 9 and 22, known as the Philadelphia chromosome, occurs in about 20% of adult and 5% in pediatric cases of ALL.
A translocation between chromosomes 4 and 11 occurs in about 4% of cases and is most common in infants under 12 months.
Not all translocations of chromosomes carry a poorer prognosis. Some translocations are relatively favorable. For example, Hyperdiploidy (>50 chromosomes) is a good prognostic factor.
Genome-wide assessment of copy number changes can be done by conventional cytogenetics or virtual karyotyping. SNP array virtual karyotyping can detect copy number changes and LOH status, while arrayCGH can detect only copy number changes. Copy neutral LOH (acquired uniparental disomy) has been reported at key loci in ALL, such as CDKN2A gene at 9p, which have prognostic significance.[53][54][55] SNP array virtual karyotyping can readily detect copy neutral LOH. Array CGH, FISH, and conventional cytogenetics cannot detect copy neutral LOH.
Correlation of prognosis with bone marrow cytogenetic finding in acute lymphoblastic leukemia
Prognosis
Cytogenetic findings
Favorable
Hyperdiploidy > 50; t (12;21)
Intermediate
Hyperdioloidy 47 -50; Normal(diploidy); del (6q); Rearrangements of 8q24
Unfavorable
Hypodiploidy-near haploidy; Near tetraploidy; del (17p); t (9;22); t (11q23)
Unclassified ALL is considered to have an intermediate prognosis.[56]
Myelodysplastic syndrome
Myelodysplastic syndrome (MDS) has remarkable clinical, morphological, and genetic heterogeneity. Cytogenetics play a decisive role in the World Health Organization's classification-based International Prognostic Scoring System (IPSS) for MDS.[57][58]
Good Prognosis: normal karyotype, isolated del(5q), isolated del(20q), -Y
Poor Prognosis: complex abnormalities (i.e., >=3 abnormalities), −7 or del(7q)
Intermediate Prognosis: all other abnormalities, including trisomy 8 and del(11q)
In a comparison of metaphase cytogenetics, FISH panel, and SNP array karyotyping for MDS, it was found that each technique provided a similar diagnostic yield. No single method detected all defects, and detection rates improved by ~5% when all three methods were used.[59]
Acquired UPD, which is not detectable by FISH or cytogenetics, has been reported at several key loci in MDS using SNP array karyotyping, including deletion of 7/7q.[60][61]
Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) including polycythemia vera, essential thrombocythemia, and primary myelofibrosis show an inherent tendency for transformation into leukemia (MPN-blast phase), which is accompanied by acquisition of additional genomic lesions. In a study of 159 cases,[62] SNP-array analysis was able to capture practically all cytogenetic abnormalities and to uncover additional lesions with potentially important clinical implications.[citation needed]
The number of genomic alterations was more than 2 to 3 times greater in the blast phase as in the chronic phase of the disease.
Deletion of 17p (TP53) was significantly associated with prior exposure to hydroxyurea as well as a complex karyotype in samples with MPN-blast crisis. Both deletion and 17p copy neutral LOH, were associated with a complex karyotype, a poor prognostic marker in myeloid malignancies. Copy neutral LOH (acquired UPD)is readily detectably by SNP array karyotype, but not by cytogenetics, FISH, or array CGH.
Blast phase patients with loss of chromosomal material on 7q showed poor survival. Loss of 7q is known to be predictive for rapid progression and poor response in AML therapy. MPN-blast phase patients with cytogenetically undetectable 7q copy neutral-LOH had comparable survival rates to those with 7/7q in their leukemic cells.
9p copy neutral-LOH with homozygous JAK2 mutation was also linked to an inferior outcome in MPN-blast crisis in comparison with patients with either heterozygous JAK2V617F or wild-type JAK2. In contrast to LOH on 17p, the prognostic impact of 9pCNN-LOH was independent of established risk factors such as 7/7q, 5q, or complex karyotype.
Colorectal cancer
Identification of biomarkers in colorectal cancer is particularly important for patients with stage II disease, where less than 20% have tumor recurrence. 18q LOH is an established biomarker associated with high risk of tumor recurrence in stage II colon cancer.[63] Figure 7 shows a SNP array karyotype of a colorectal carcinoma (whole genome view).
Colorectal cancers are classified into specific tumor phenotypes based on molecular profiles[63] which can be integrated with the results of other ancillary tests, such as microsatellite instability testing, IHC, and KRAS mutation status:
Chromosomal instability (CIN) which have allelic imbalance at a number of chromosomal loci, including 5q, 8p, 17p, and 18q (Fig 7).
Microsatellite instability (MSI) which tend to have diploid karyotypes.
Malignant rhabdoid tumors
Malignant rhabdoid tumors are rare, highly aggressive neoplasms found most commonly in infants and young children. Due to their heterogenous histologic features, diagnosis can often be difficult and misclassifications can occur. In these tumors, the INI1 gene (SMARCB1)on chromosome 22q functions as a classic tumor suppressor gene. Inactivation of INI1 can occur via deletion, mutation, or acquired UPD.[64]
In a recent study,[64] SNP array karyotyping identified deletions or LOH of 22q in 49/51 rhabdoid tumors. Of these, 14 were copy neutral LOH (or acquired UPD), which is detectable by SNP array karyotyping, but not by FISH, cytogenetics, or arrayCGH. MLPA detected a single exon homozygous deletion in one sample that was below the resolution of the SNP array.[citation needed]
SNP array karyotyping can be used to distinguish, for example, a medulloblastoma with an isochromosome 17q from a primary rhabdoid tumor with loss of 22q11.2. When indicated, molecular analysis of INI1 using MLPA and direct sequencing may then be employed. Once the tumor-associated changes are found, an analysis of germline DNA from the patient and the parents can be done to rule out an inherited or de novo germline mutation or deletion of INI1, so that appropriate recurrence risk assessments can be made.[64]
Uveal melanoma
The most important genetic alteration associated with poor prognosis in uveal melanoma is loss of an entire copy of Chromosome 3 (Monosomy 3), which is strongly correlated with metastatic spread.[65] Gains on chromosomes 6 and 8 are often used to refine the predictive value of the Monosomy 3 screen, with gain of 6p indicating a better prognosis and gain of 8q indicating a worse prognosis in disomy 3 tumors.[66] In rare instances, monosomy 3 tumors may duplicate the remaining copy of the chromosome to return to a disomic state referred to as isodisomy.[67] Isodisomy 3 is prognostically equivalent to monosomy 3, and both can be detected by tests for chromosome 3 loss of heterozygosity.[68]
Limitations
Unlike karyotypes obtained from conventional cytogenetics, virtual karyotypes are reconstructed by computer programs using signals obtained from disrupted DNA. In essence, the computer program will correct translocations when it lines up the signals in chromosomal order. Therefore, virtual karyotypes cannot detect balanced translocations and inversions. They also can only detect genetic aberrations in regions of the genome that are represented by probes on the array. In addition, virtual karyotypes generate a relative copy number normalized against a diploid genome, so tetraploid genomes will be condensed into a diploid space unless renormalization is performed. Renormalization requires an ancillary cell-based assay, such as FISH, if one is using arrayCGH. For karyotypes obtained from SNP-based arrays, tetraploidy can often be inferred from the maintenance of heterozygosity within a region of apparent copy number loss.[22] Low-level mosaicism or small subclones may not be detected by virtual karyotypes because the presence of normal cells in the sample will dampen the signal from the abnormal clone. The exact point of failure, in terms of the minimal percentage of neoplastic cells, will depend on the particular platform and algorithms used. Many copy number analysis software programs used to generate array-based karyotypes will falter with less than 25–30% tumor/abnormal cells in the sample. However, in oncology applications this limitation can be minimized by tumor enrichment strategies and software optimized for use with oncology samples. The analysis algorithms are evolving rapidly, and some are even designed to thrive on 'normal clone contamination',[69] so it is anticipated that this limitation will continue to dissipate.
See also
DECIPHER, a Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources
↑ Kulharya AS, Flannery DB, Norris K, Lovell C, Levy B, Velagaleti G (September 2008). "Fine mapping of breakpoints in two unrelated patients with rare overlapping interstitial deletions of 9q with mild dysmorphic features". American Journal of Medical Genetics. 146A (17): 2234–41. doi:10.1002/ajmg.a.32397. PMID18666229. S2CID25455126.
↑ Nowakowska B; Stankiewicz P; Obersztyn E; Ou Z; Li J; Chinault AC; Smyk M; Borg K; Mazurczak T; Cheung SW; Bocian E (September 2008). "Application of metaphase HR-CGH and targeted Chromosomal Microarray Analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features". American Journal of Medical Genetics. 146A (18): 2361–9. doi:10.1002/ajmg.a.32475. PMID18698622. S2CID30882747.
↑ Probst FJ; Roeder ER; Enciso VB; Ou Z; Cooper ML; Eng P; Li J; Gu Y; Stratton RF; Chinault AC; Shaw CA; Sutton VR; Cheung SW; Nelson DL (June 2007). "Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation". American Journal of Medical Genetics. 143A (12): 1358–65. doi:10.1002/ajmg.a.31781. PMID17506108. S2CID22090841.
↑ Ishikawa S; Komura D; Tsuji S; Nishimura K; Yamamoto S; Panda B; Huang J; Fukayama M; Jones KW; Aburatani H (August 2005). "Allelic dosage analysis with genotyping microarrays". Biochem Biophys Res Commun. 333 (4): 1309–14. doi:10.1016/j.bbrc.2005.06.040. PMID15982637.
1 2 Lo KC, Bailey D, Burkhardt T, Gardina P, Turpaz Y, Cowell J (March 2008). "Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization". Genes Chromosomes Cancer. 47 (3): 221–37. doi:10.1002/gcc.20524. PMID18050302. S2CID19480318.
↑ Michels E, Vandesompele J, Hoebeeck J, Menten B, De Preter K, Laureys G, Van Roy N, Speleman F (2006). "Genome wide measurement of DNA copy number changes in neuroblastoma: dissecting amplicons and mapping losses, gains and breakpoints". Cytogenet. Genome Res. 115 (3–4): 273–282. doi:10.1159/000095924. PMID17124410. S2CID14012430.
↑ Messahel B; Williams R; Ridolfi A; A'hern R; Warren W; Tinworth L; Hobson R; Al-Saadi R; Whyman G; Brundler MA; Kelsey A; Sebire N; Jones C; Vujanic G; Pritchard-Jones K; Children's Cancer and Leukaemia Group (CCLG) (March 2009). "Children's Cancer and Leukaemia Group (CCLG). Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study". Eur J Cancer. 45 (5): 819–26. doi:10.1016/j.ejca.2009.01.005. PMID19231157.
↑ White VA, McNeil BK, Horsman DE (1998). "Acquired homozygosity (isodisomy) of chromosome 3 in uveal melanoma". Cancer Genet Cytogenet. 102 (1): 40–45. doi:10.1016/S0165-4608(97)00290-2. PMID9530338.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.