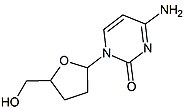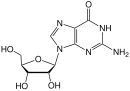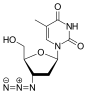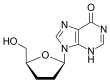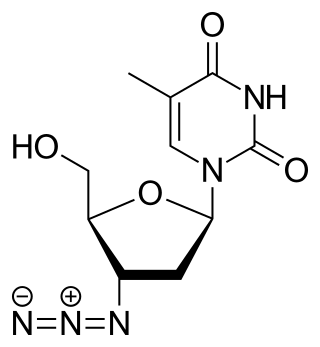
Zidovudine (ZDV), also known as azidothymidine (AZT), was the first antiretroviral medication used to prevent and treat HIV/AIDS. It is generally recommended for use in combination with other antiretrovirals. It may be used to prevent mother-to-child spread during birth or after a needlestick injury or other potential exposure. It is sold both by itself and together as lamivudine/zidovudine and abacavir/lamivudine/zidovudine. It can be used by mouth or by slow injection into a vein.
The management of HIV/AIDS normally includes the use of multiple antiretroviral drugs as a strategy to control HIV infection. There are several classes of antiretroviral agents that act on different stages of the HIV life-cycle. The use of multiple drugs that act on different viral targets is known as highly active antiretroviral therapy (HAART). HAART decreases the patient's total burden of HIV, maintains function of the immune system, and prevents opportunistic infections that often lead to death. HAART also prevents the transmission of HIV between serodiscordant same-sex and opposite-sex partners so long as the HIV-positive partner maintains an undetectable viral load.
Reverse-transcriptase inhibitors (RTIs) are a class of antiretroviral drugs used to treat HIV infection or AIDS, and in some cases hepatitis B. RTIs inhibit activity of reverse transcriptase, a viral DNA polymerase that is required for replication of HIV and other retroviruses.

Didanosine, sold under the brand name Videx, is a medication used to treat HIV/AIDS. It is used in combination with other medications as part of highly active antiretroviral therapy (HAART). It is of the reverse-transcriptase inhibitor class.

Zalcitabine, also called dideoxycytidine, is a nucleoside analog reverse-transcriptase inhibitor (NRTI) sold under the trade name Hivid. Zalcitabine was the third antiretroviral to be approved by the Food and Drug Administration (FDA) for the treatment of HIV/AIDS. It is used as part of a combination regimen.

Stavudine (d4T), sold under the brand name Zerit among others, is an antiretroviral medication used to prevent and treat HIV/AIDS. It is generally recommended for use with other antiretrovirals. It may be used for prevention after a needlestick injury or other potential exposure. However, it is not a first-line treatment. It is given by mouth.

Lamivudine, commonly called 3TC, is an antiretroviral medication used to prevent and treat HIV/AIDS. It is also used to treat chronic hepatitis B when other options are not possible. It is effective against both HIV-1 and HIV-2. It is typically used in combination with other antiretrovirals such as zidovudine, dolutegravir, and abacavir. Lamivudine may be included as part of post-exposure prevention in those who have been potentially exposed to HIV. Lamivudine is taken by mouth as a liquid or tablet.

Abacavir, sold under the brand name Ziagen among others, is a medication used to treat HIV/AIDS. Similar to other nucleoside analog reverse-transcriptase inhibitors (NRTIs), abacavir is used together with other HIV medications, and is not recommended by itself. It is taken by mouth as a tablet or solution and may be used in children over the age of three months.
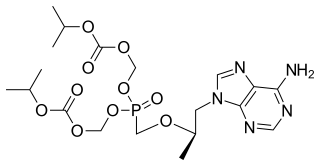
Tenofovir disoproxil, sold under the trade name Viread among others, is a medication used to treat chronic hepatitis B and to prevent and treat HIV/AIDS. It is generally recommended for use with other antiretrovirals. It may be used for prevention of HIV/AIDS among those at high risk before exposure, and after a needlestick injury or other potential exposure. It is sold both by itself and together in combinations such as emtricitabine/tenofovir, efavirenz/emtricitabine/tenofovir, and elvitegravir/cobicistat/emtricitabine/tenofovir. It does not cure HIV/AIDS or hepatitis B. It is available by mouth as a tablet or powder.

Nevirapine (NVP), sold under the brand name Viramune among others, is a medication used to treat and prevent HIV/AIDS, specifically HIV-1. It is generally recommended for use with other antiretroviral medications. It may be used to prevent mother to child spread during birth but is not recommended following other exposures. It is taken by mouth.

Entecavir (ETV), sold under the brand name Baraclude, is an antiviral medication used in the treatment of hepatitis B virus (HBV) infection. In those with both HIV/AIDS and HBV antiretroviral medication should also be used. Entecavir is taken by mouth as a tablet or solution.
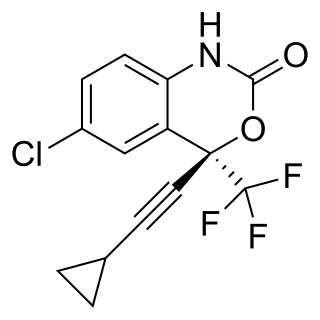
Efavirenz (EFV), sold under the brand names Sustiva among others, is an antiretroviral medication used to treat and prevent HIV/AIDS. It is generally recommended for use with other antiretrovirals. It may be used for prevention after a needlestick injury or other potential exposure. It is sold both by itself and in combination as efavirenz/emtricitabine/tenofovir. It is taken by mouth.

Nucleoside analogues are structural analogues of a nucleoside, which normally contain a nucleobase and a sugar. Nucleotide analogues are analogues of a nucleotide, which normally has one to three phosphates linked to a nucleoside. Both types of compounds can deviate from what they mimick in a number of ways, as changes can be made to any of the constituent parts. They are related to nucleic acid analogues.

Lamivudine/zidovudine, sold under the brand name Combivir among others, is a fixed-dose combination antiretroviral medication used to treat HIV/AIDS. It contains two antiretroviral medications, lamivudine and zidovudine. It is used together with other antiretrovirals. It is taken by mouth twice a day.

A resistance mutation is a mutation in a virus gene that allows the virus to become resistant to treatment with a particular antiviral drug. The term was first used in the management of HIV, the first virus in which genome sequencing was routinely used to look for drug resistance. At the time of infection, a virus will infect and begin to replicate within a preliminary cell. As subsequent cells are infected, random mutations will occur in the viral genome. When these mutations begin to accumulate, antiviral methods will kill the wild type strain, but will not be able to kill one or many mutated forms of the original virus. At this point a resistance mutation has occurred because the new strain of virus is now resistant to the antiviral treatment that would have killed the original virus. Resistance mutations are evident and widely studied in HIV due to its high rate of mutation and prevalence in the general population. Resistance mutation is now studied in bacteriology and parasitology.

Rilpivirine, sold under the brand names Edurant and Rekambys, is a medication, developed by Tibotec, used for the treatment of HIV/AIDS. It is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with higher potency, longer half-life and reduced side-effect profile compared with older NNRTIs such as efavirenz.
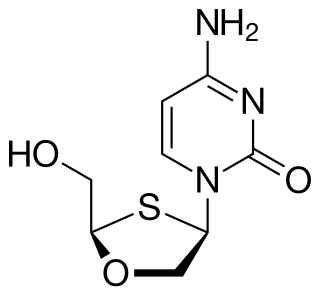
Apricitabine is an experimental nucleoside reverse transcriptase inhibitor (NRTI) against HIV. It is structurally related to lamivudine and emtricitabine, and, like these, is an analogue of cytidine.

Elvucitabine is an experimental nucleoside reverse transcriptase inhibitor (NRTI), developed by Achillion Pharmaceuticals, Inc. for the treatment of HIV infection.
Non-nucleoside reverse-transcriptase inhibitors (NNRTIs) are antiretroviral drugs used in the treatment of human immunodeficiency virus (HIV). NNRTIs inhibit reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of HIV. RT is one of the most popular targets in the field of antiretroviral drug development.

Bictegravir/emtricitabine/tenofovir alafenamide, sold under the brand name Biktarvy, is a fixed-dose combination antiretroviral medication for the treatment of HIV/AIDS. One tablet, taken orally once daily, contains 50 mg bictegravir, 200 mg emtricitabine, and 25 mg tenofovir alafenamide. It was approved for use in the United States in February 2018, and for use in the European Union in June 2018.