
A phosphodiesterase inhibitor is a drug that blocks one or more of the five subtypes of the enzyme phosphodiesterase (PDE), thereby preventing the inactivation of the intracellular second messengers, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) by the respective PDE subtype(s). The ubiquitous presence of this enzyme means that non-specific inhibitors have a wide range of actions, the actions in the heart, and lungs being some of the first to find a therapeutic use.

Sildenafil, sold under the brand name Viagra, among others, is a medication used to treat erectile dysfunction and pulmonary arterial hypertension. It is also sometimes used off-label for the treatment of certain symptoms in secondary Raynaud's phenomenon. It is unclear if it is effective for treating sexual dysfunction in women. It can be taken orally, intravenously, or sublingually. Onset when taken orally is typically within twenty minutes and lasts for about two hours.
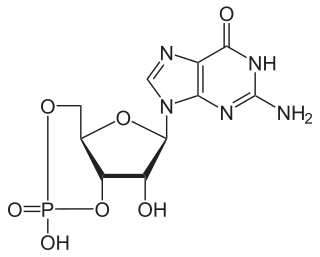
A phosphodiesterase (PDE) is an enzyme that breaks a phosphodiester bond. Usually, phosphodiesterase refers to cyclic nucleotide phosphodiesterases, which have great clinical significance and are described below. However, there are many other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases, as well as numerous less-well-characterized small-molecule phosphodiesterases.

Tadalafil, sold under the brand name Cialis among others, is a medication used to treat erectile dysfunction, benign prostatic hyperplasia, and pulmonary arterial hypertension. It is taken by mouth. Onset is typically within half an hour and the duration is up to 36 hours.

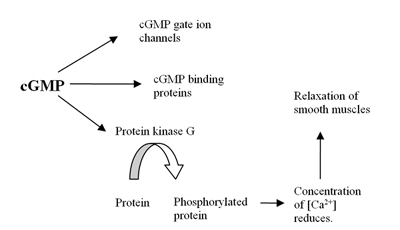
Cyclic guanosine monophosphate (cGMP) is a cyclic nucleotide derived from guanosine triphosphate (GTP). cGMP acts as a second messenger much like cyclic AMP. Its most likely mechanism of action is activation of intracellular protein kinases in response to the binding of membrane-impermeable peptide hormones to the external cell surface. Through protein kinases activation, cGMP can relax smooth muscle. cGMP concentration in urine can be measured for kidney function and diabetes detection.
Vardenafil, sold under the brand name Levitra among others, is a medication that is used for treating erectile dysfunction. It is a PDE5 inhibitor. It is taken by mouth.

A phosphodiesterase type 5 inhibitor is a vasodilating drug that works by blocking the degradative action of cGMP-specific phosphodiesterase type 5 (PDE5) on cyclic GMP in the smooth muscle cells lining the blood vessels supplying various tissues. These drugs dilate the corpora cavernosa of the penis, facilitating erection with sexual stimulation, and are used in the treatment of erectile dysfunction (ED). Sildenafil was the first effective oral treatment available for ED. Because PDE5 is also present in the smooth muscle of the walls of the arterioles within the lungs, two PDE5 inhibitors, sildenafil and tadalafil, are FDA-approved for the treatment of pulmonary hypertension. As of 2019, the wider cardiovascular benefits of PDE5 inhibitors are being appreciated.
Cyclic guanosine monophosphate-specific phosphodiesterase type 5 is an enzyme from the phosphodiesterase class. It is found in various tissues, most prominently the corpus cavernosum and the retina. It has also been recently discovered to play a vital role in the cardiovascular system.

PDE3 is a phosphodiesterase. The PDEs belong to at least eleven related gene families, which are different in their primary structure, substrate affinity, responses to effectors, and regulation mechanism. Most of the PDE families are composed of more than one gene. PDE3 is clinically significant because of its role in regulating heart muscle, vascular smooth muscle and platelet aggregation. PDE3 inhibitors have been developed as pharmaceuticals, but their use is limited by arrhythmic effects and they can increase mortality in some applications.
Phosphodiesterase 1, PDE1, EC 3.1.4.1, systematic name oligonucleotide 5′-nucleotidohydrolase) is a phosphodiesterase enzyme also known as calcium- and calmodulin-dependent phosphodiesterase. It is one of the 11 families of phosphodiesterase (PDE1-PDE11). Phosphodiesterase 1 has three subtypes, PDE1A, PDE1B and PDE1C which divide further into various isoforms. The various isoforms exhibit different affinities for cAMP and cGMP.

The PDE2 enzyme is one of 21 different phosphodiesterases (PDE) found in mammals. These different PDEs can be subdivided to 11 families. The different PDEs of the same family are functionally related despite the fact that their amino acid sequences show considerable divergence. The PDEs have different substrate specificities. Some are cAMP selective hydrolases, others are cGMP selective hydrolases and the rest can hydrolyse both cAMP and cGMP.

The helicine arteries of penis are arteries in the penis. They are found in the corpora cavernosa penis.

The drug udenafil is marketed under the trade name Zydena. It is within the PDE5 inhibitor class (which also includes avanafil, sildenafil, tadalafil, and vardenafil). Like other PDE5 inhibitors, it is used to treat erectile dysfunction. Udenafil was developed by Dong-A Pharmaceutical. It has fairly rapid onset of action (peak plasma concentration after 1 to 1.5 hours), and has long duration of action (plasma half-life of 11 to 13 hours). Udenafil's pharmacokinetics allows once-daily dosage (in addition to on-demand use). Typical doses are 100 and 200 mg. Udenafil is available in Korea, Russia, and the Philippines. It has not yet been approved for use in the United States by the U.S. Food and Drug Administration.

Avanafil is a PDE5 inhibitor approved for erectile dysfunction by the FDA on April 27, 2012 and by EMA on June 21, 2013. Avanafil is sold under the brand names Stendra and Spedra. It was invented at Mitsubishi Tanabe Pharma, formerly known as Tanabe Seiyaku Co., and licensed to Vivus Inc., which partnered with Menarini Group to commercialise Spedra in over forty European countries, Australia, and New Zealand. Metuchen Pharmaceuticals obtained exclusive rights within the United States.
Acetildenafil (hongdenafil) is a synthetic drug which acts as a phosphodiesterase inhibitor. It is an analog of sildenafil (Viagra) which has been detected in numerous different brands of supposedly "herbal" aphrodisiac products sold to boost libido and alleviate erectile dysfunction.

Lodenafil is a drug belonging to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. Like udenafil and avanafil it belongs to a new generation of PDE5 inhibitors.

Mirodenafil belongs to the drug class PDE5 inhibitors, which includes avanafil, sildenafil, tadalafil, udenafil, and vardenafil, and is the first-line treatment for erectile dysfunction. Developed by SK Chemicals Life Science, mirodenafil is marketed in Korea under the trade name Mvix, offered both as tablets and as orally dissolving film.

Sulfoaildenafil (thioaildenafil) is a synthetic drug that is a structural analog of sildenafil (Viagra). It was first reported in 2005, and it is not approved by any health regulation agency. Like sildenafil, sulfoaildenafil is a phosphodiesterase type 5 inhibitor.

Homosildenafil is a synthetic drug which acts as a phosphodiesterase inhibitor. It is an analog of sildenafil and vardenafil. Homosildenafil was first identified as an adulterant in sex enhancement products in 2003 and was more recently detected in dietary supplements.

Ototoxicity is defined as the toxic effect on the functioning of the inner ear, which may lead to temporary or permanent hearing loss (cochleotoxic) and balancing problems (vestibulotoxic). Drugs or pharmaceutical agents inducing ototoxicity are regarded as ototoxic medications.










{kind=link}
{kind=link}