
Zolpidem, sold under the brand name Ambien among others, is a medication primarily used for the short-term treatment of sleeping problems. Guidelines recommend that it be used only after cognitive behavioral therapy for insomnia and behavioral changes, such as sleep hygiene, have been tried. It decreases the time to sleep onset by about fifteen minutes and at larger doses helps people stay asleep longer. It is taken by mouth and is available in conventional tablets, sublingual tablets, or oral spray.

An imidazopyridine is a nitrogen containing heterocycle that is also a class of drugs that contain this same chemical substructure. In general, they are GABAA receptor agonists, however recently proton pump inhibitors, aromatase inhibitors, NSAIDs and other classes of drugs in this class have been developed as well. Despite usually being similar to them in effect, they are not chemically related to benzodiazepines. As such, GABAA-agonizing imidazopyridines, pyrazolopyrimidines, and cyclopyrrones are sometimes grouped together and referred to as "nonbenzodiazepines." Imidazopyridines include:

The GABAA receptor (GABAAR) is an ionotropic receptor and ligand-gated ion channel. Its endogenous ligand is γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system.Accurate regulation of GABAergic transmission through appropriate developmental processes, specificity to neural cell types, and responsiveness to activity is crucial for the proper functioning of nearly all aspects of the central nervous system (CNS). Upon opening, the GABAA receptor on the postsynaptic cell is selectively permeable to chloride ions (Cl−) and, to a lesser extent, bicarbonate ions (HCO3−). Depending on the membrane potential and the ionic concentration difference, this can result in ionic fluxes across the pore. If the membrane potential is higher than the equilibrium potential (also known as the reversal potential) for chloride ions, when the receptor is activated Cl− will flow into the cell. This causes an inhibitory effect on neurotransmission by diminishing the chance of a successful action potential occurring at the postsynaptic cell. The reversal potential of the GABAA-mediated inhibitory postsynaptic potential (IPSP) in normal solution is −70 mV, contrasting the GABAB IPSP (-100 mV).
Nonbenzodiazepines, sometimes referred to colloquially as Z-drugs, are a class of psychoactive drugs that are benzodiazepine-like in uses, such as for treating insomnia and anxiety.
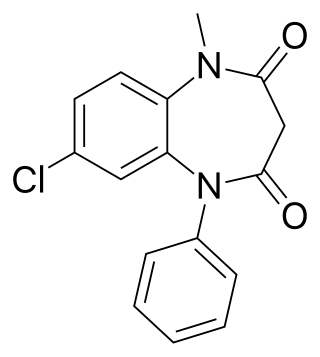
Clobazam, sold under the brand names Frisium, Onfi and others, is a benzodiazepine class medication that was patented in 1968. Clobazam was first synthesized in 1966 and first published in 1969. Clobazam was originally marketed as an anxioselective anxiolytic since 1970, and an anticonvulsant since 1984. The primary drug-development goal was to provide greater anxiolytic, anti-obsessive efficacy with fewer benzodiazepine-related side effects.

Bretazenil (Ro16-6028) is an imidazopyrrolobenzodiazepine anxiolytic drug which is derived from the benzodiazepine family, and was invented in 1988. It is most closely related in structure to the GABA antagonist flumazenil, although its effects are somewhat different. It is classified as a high-potency benzodiazepine due to its high affinity binding to benzodiazepine binding sites where it acts as a partial agonist. Its profile as a partial agonist and preclinical trial data suggests that it may have a reduced adverse effect profile. In particular bretazenil has been proposed to cause a less strong development of tolerance and withdrawal syndrome. Bretazenil differs from traditional 1,4-benzodiazepines by being a partial agonist and because it binds to α1, α2, α3, α4, α5 and α6 subunit containing GABAA receptor benzodiazepine receptor complexes. 1,4-benzodiazepines bind only to α1, α2, α3 and α5GABAA benzodiazepine receptor complexes.

Pagoclone is an anxiolytic agent from the cyclopyrrolone family, related to better-known drugs such as the sleeping medication zopiclone. It was synthesized by a French team working for Rhone-Poulenc & Rorer S.A. Pagoclone belongs to the class of nonbenzodiazepines, which have similar effects to the older benzodiazepine group, but with quite different chemical structures. It was never commercialised.

Etifoxine, sold under the trade name Stresam among others, is a nonbenzodiazepine anxiolytic agent, primarily indicated for short-term management of adjustment disorder, specifically instances of situational depression accompanied by anxiety, such as stress-induced anxiety. Administration is by mouth. Side effects associated with etifoxine use include slight drowsiness, headache, skin eruptions, and allergic reactions. In rare cases, etifoxine has been linked to severe skin and liver toxicity, as well as menstrual bleeding between periods. Unlike benzodiazepines, etifoxine does not cause sedation or lack of coordination. Etifoxine acts as a GABAA receptor positive allosteric modulator and as a ligand for translocator proteins. Both mechanisms are conjectured to contribute to its anxiolytic properties.

Ocinaplon is an anxiolytic drug in the pyrazolopyrimidine family of drugs. Other pyrazolopyrimidine drugs include zaleplon and indiplon.
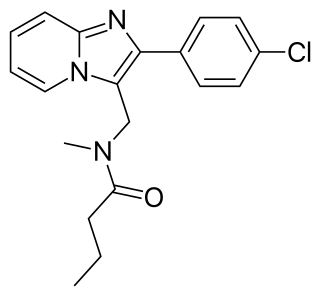
Saripidem is a sedative and anxiolytic drug in the imidazopyridine family, which is related to the better known drugs zolpidem and alpidem.
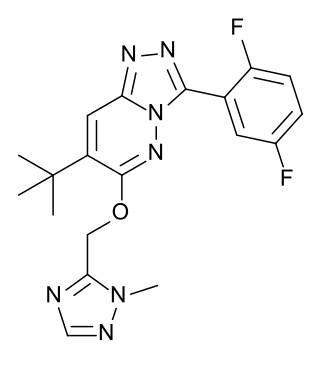
L-838,417 is an anxiolytic drug used in scientific research. It has similar effects to benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine anxiolytic. The compound was developed by Merck, Sharp and Dohme.

SL651498 is an anxiolytic and anticonvulsant drug used in scientific research, with a chemical structure most closely related to β-carboline derivatives such as abecarnil and gedocarnil. It has similar effects to benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine anxiolytic.

CL-218,872 is a sedative and hypnotic drug used in scientific research. It has similar effects to sedative-hypnotic benzodiazepine drugs such as triazolam, but is structurally distinct and so is classed as a nonbenzodiazepine hypnotic.

SX-3228 is a sedative and hypnotic drug used in scientific research. It has similar effects to sedative-hypnotic benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine hypnotic.

Tracazolate (ICI-136,753) is an anxiolytic drug which is used in scientific research. It is a pyrazolopyridine derivative, most closely related to pyrazolopyrimidine drugs such as zaleplon, and is one of a structurally diverse group of drugs known as the nonbenzodiazepines which act at the same receptor targets as benzodiazepines but have distinct chemical structures.

TPA-023 (MK-0777) is an anxiolytic drug with a novel chemical structure, which is used in scientific research. It has similar effects to benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine anxiolytic. It is a subtype-selective, mixed allosteric modular at the benzodiazepine location on GABAA receptors, where it acts as a partial agonist at the α2 and α3 subtypes, but as a silent antagonist at α1 and α5 subtypes. It has primarily anxiolytic and anticonvulsant effects in animal tests, but with no sedative effects even at 50 times the effective anxiolytic dose.

L-655,708 (FG-8094) is a nootropic drug invented in 1996 by a team working for Merck, Sharp and Dohme, that was the first compound developed which acts as a subtype-selective inverse agonist at the α5 subtype of the benzodiazepine binding site on the GABAA receptor. It acts as an inverse agonist at the α1, α2, α3 and α5 subtypes, but with much higher affinity for α5, and unlike newer α5 inverse agonists such as α5IA, L-655,708 exerts its subtype selectivity purely via higher binding affinity for this receptor subtype, with its efficacy as an inverse agonist being around the same at all the subtypes it binds to.

DS-1 is a drug from the imidazopyridine family, which is the first drug developed that acts as a GABAA receptor positive allosteric modulator (PAM) selective for the α4β3δ subtype, which is not targeted by other GABAA receptor PAMs such as the benzodiazepines or other nonbenzodiazepine drugs. Novel selective drugs such as DS-1 should prove useful in the study of this receptor subtype.
A GABAA receptor negative allosteric modulator is a negative allosteric modulator (NAM), or inhibitor, of the GABAA receptor, a ligand-gated ion channel of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA). They are closely related and similar to GABAA receptor antagonists. The effects of GABAA receptor NAMs are functionally the opposite of those of GABAA receptor positive allosteric modulators (PAMs) like the benzodiazepines, barbiturates, and ethanol (alcohol). Non-selective GABAA receptor NAMs can produce a variety of effects including convulsions, neurotoxicity, and anxiety, among others.

Darigabat is a GABAergic medication which is under development for the treatment of photosensitive epilepsy, focal onset seizures, panic disorder, and other anxiety disorders. It was also under development for the treatment of generalized anxiety disorder and chronic lower back pain, but development for these indications was discontinued. It is taken via oral administration.
