
Harlequin-type ichthyosis is a genetic disorder that results in thickened skin over nearly the entire body at birth. The skin forms large, diamond/trapezoid/rectangle-shaped plates that are separated by deep cracks. These affect the shape of the eyelids, nose, mouth, and ears and limit movement of the arms and legs. Restricted movement of the chest can lead to breathing difficulties. These plates fall off over several weeks. Other complications can include premature birth, infection, problems with body temperature, and dehydration. The condition is the most severe form of ichthyosis, a group of genetic disorders characterised by scaly skin.

Incontinentia pigmenti (IP) is a rare X-linked dominant genetic disorder that affects the skin, hair, teeth, nails and central nervous system. It is named from its appearance under a microscope.

Bloom syndrome is a rare autosomal recessive genetic disorder characterized by short stature, predisposition to the development of cancer, and genomic instability. BS is caused by mutations in the BLM gene which is a member of the RecQ DNA helicase family. Mutations in genes encoding other members of this family, namely WRN and RECQL4, are associated with the clinical entities Werner syndrome and Rothmund–Thomson syndrome, respectively. More broadly, Bloom syndrome is a member of a class of clinical entities that are characterized by chromosomal instability, genomic instability, or both and by cancer predisposition.
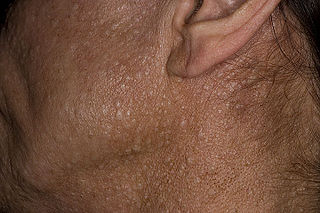
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons is a human, adult onset, autosomal dominant genetic disorder caused by a mutation in the folliculin (FLCN) gene. It can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%) and 24% of people with BHD eventually experience a collapsed lung. Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.

X-linked ichthyosis is a skin condition caused by the hereditary deficiency of the steroid sulfatase (STS) enzyme that affects 1 in 2000 to 1 in 6000 males. XLI manifests with dry, scaly skin and is due to deletions or mutations in the STS gene. XLI can also occur in the context of larger deletions causing contiguous gene syndromes. Treatment is largely aimed at alleviating the skin symptoms. The term is from the Ancient Greek 'ichthys' meaning 'fish'.

Meleda disease (MDM) or "mal de Meleda", also called Mljet disease, keratosis palmoplantaris and transgradiens of Siemens, is an extremely rare autosomal recessive congenital skin disorder in which dry, thick patches of skin develop on the soles of the hands and feet, a condition known as palmoplantar hyperkeratosis. Meleda Disease is a skin condition which usually can be identified not long after birth. This is a genetic condition but it is very rare. The hands and feet usually are the first to show signs of the disease but the disease can advance to other parts of the body. Signs of the disease include thickening of the skin, on hands and soles of feet, which can turn red in color. There currently is no cure and treatment is limited, but Acitretin can be used in severe cases.
Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and MDS/AML, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing and cognitive impairment can be a feature. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.
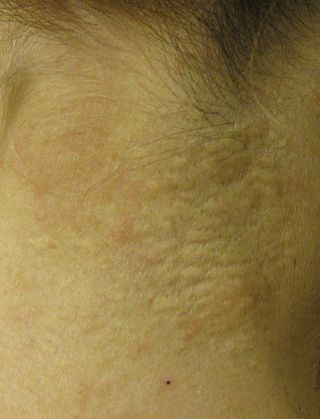
Pseudoxanthoma elasticum (PXE) is a genetic disease that causes mineralization of elastic fibers in some tissues. The most common problems arise in the skin and eyes, and later in blood vessels in the form of premature atherosclerosis. PXE is caused by autosomal recessive mutations in the ABCC6 gene on the short arm of chromosome 16 (16p13.1).
Conradi–Hünermann syndrome is a rare type of chondrodysplasia punctata. It is associated with the EBP gene and affects between one in 100,000 and one in 200,000 babies.

Pachyonychia congenita is a rare group of autosomal dominant skin disorders that are caused by a mutation in one of five different keratin genes. Pachyonychia congenita is often associated with thickened toenails, plantar keratoderma, and plantar pain.
Ablepharon macrostomia syndrome (AMS) is an extremely rare, autosomal dominant genetic disorder characterized by abnormal phenotypic appearances that primarily affect the head and face as well as the skull, skin, fingers and genitals. AMS generally results in abnormal ectoderm-derived structures. The most prominent abnormality is the underdevelopment (microblepharon) or absence of eyelids – signifying the ablepharon aspect of the disease – and a wide, fish-like mouth – macrostomia. Recent scholars and surgeons have called into question the naming of the condition as "Ablepharon" on account of recent investigation and histology showing consistent evidence of at least some eyelid tissue. Infants presenting with AMS may also have malformations of the abdominal wall and nipples. Children with AMS might also experience issues with learning development, language difficulties and intellectual disabilities.

Ichthyosis bullosa of Siemens is a type of familial, autosomal dominant ichthyosis, a rare skin disorder. It is also known as bullous congenital ichthyosiform erythroderma of Siemens or ichthyosis exfoliativa. It is a genetic disorder with no known cure which is estimated to affect about 1 in 500,000 people.

Urbach–Wiethe disease is a very rare recessive genetic disorder, with approximately 400 reported cases since its discovery. It was first officially reported in 1929 by Erich Urbach and Camillo Wiethe, although cases may be recognized dating back as early as 1908.

Warty dyskeratoma, also known as an Isolated dyskeratosis follicularis, is a benign epidermal proliferation with distinctive histologic findings that may mimic invasive squamous cell carcinoma and commonly manifests as an umbilicated lesion with a keratotic plug, WD have some histopathologic similarities to viral warts but it's not caused by HPV and the majority of these lesions display overall histopathologic features consistent with a follicular adnexal neoplasm. Usually limited to the head, neck, scalp or face and vulva. Lesions are generally solitary and sporadic and may be associated with a follicular unit. Oral involvement, particularly the hard palate, and genital involvement have been reported. it can also be thought of as one of the manifestations of focal acantholytic dyskeratosis, an epidermal reaction pattern that can be seen in several disorders, including Darier's disease and Grover's disease. But the main Difference between Darier disease and Warty dyskeratoma, is that Darier disease inherited dermatosis consisting of multiple keratotic papules on the face, trunk, and extremities, while WD occurs as an isolated, noninherited, single keratotic nodule mainly confined to the head and neck as mentioned earlier.
Acrokeratosis verruciformis is a rare autosomal dominant disorder appearing at birth or in early childhood, characterized by skin lesions that are small, verrucous, flat papules resembling warts along with palmoplantar punctate keratoses and pits. However sporadic forms, whose less than 10 cases have been reported, presents at a later age, usually after the first decade and generally lack palmoplantar keratoses. Whether acrokeratosis verruciformis and Darier disease are related or distinct entities has been controversial, like Darier's disease, it is associated with defects in the ATP2A2 gene. however the specific mutations found in the ATP2A2 gene in acrokeratosis verruciformis have never been found in Darier's disease.
Wrinkly skin syndrome(WSS) is a rare genetic condition characterized by sagging, wrinkled skin, low skin elasticity, and delayed fontanelle (soft spot) closure, along with a range of other symptoms. The disorder exhibits an autosomal recessive inheritance pattern with mutations in the ATP6V0A2 gene, leading to abnormal glycosylation events. There are only about 30 known cases of WSS as of 2010. Given its rarity and symptom overlap with other dermatological conditions, reaching an accurate diagnosis is difficult and requires specialized dermatological testing. Limited treatment options are available but long-term prognosis is variable from patient to patient, based on individual case studies. Some skin symptoms recede with increasing age, while progressive neurological advancement of the disorder causes seizures and mental deterioration later in life for some patients.
Ichthyosis prematurity syndrome (IPS) is a dermatological disease with known genetic causes. This syndrome is a rare subcategory of autosomal recessive congenital ichthyosis (ARCI). It is associated with complications in the mid-trimester of a pregnancy leading to premature births. Although most prevalent in individuals of Scandinavian origin, there have also been scattered cases in people of Japanese, Italian and Indian ethnicity. This disorder is also referred to as ichthyosis congenital type IV.
Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.
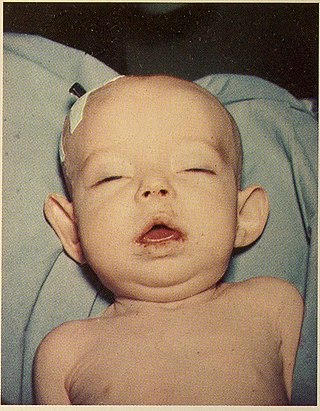
Donohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
Familial disseminated comedones without dyskeratosis (FDCWD) is a rare autosomal dominant skin disorder characterized by the presence of numerous comedones on the face, trunk, and extremities. The comedones are typically asymptomatic and do not lead to scarring. The disorder is thought to be caused by a mutation in the gene encoding the protein involucrin.



