
Salvinorin A is the main active psychotropic molecule in Salvia divinorum. Salvinorin A is considered a dissociative hallucinogen.
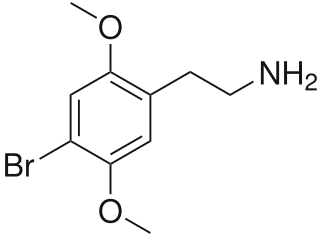
2C-B (4-Bromo-2,5-dimethoxyphenethylamine) is a psychedelic drug of the 2C family. It was first synthesized by Alexander Shulgin in 1974. In Shulgin's book PiHKAL, the dosage range is listed as 12–24 mg. As a recreational drug, 2C-B is sold as a white powder sometimes pressed in tablets or gel caps. It is also referred to by a number of street names. The drug is usually taken orally, but can also be insufflated or vaporized. While being primarily a psychedelic it is also a mild entactogen.
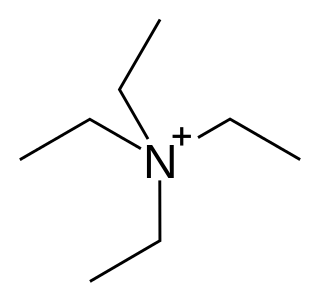
Tetraethylammonium (TEA) is a quaternary ammonium cation with the chemical formula [Et4N]+, consisting of four ethyl groups attached to a central nitrogen atom. It is a counterion used in the research laboratory to prepare lipophilic salts of inorganic anions. It is used similarly to tetrabutylammonium, the difference being that its salts are less lipophilic, more easily crystallized and more toxic.

Synephrine, or, more specifically, p-synephrine, is an alkaloid, occurring naturally in some plants and animals, and also in approved drugs products as its m-substituted analog known as neo-synephrine. p-Synephrine and m-synephrine are known for their longer acting adrenergic effects compared to epinephrine and norepinephrine. This substance is present at very low concentrations in common foodstuffs such as orange juice and other orange products, both of the "sweet" and "bitter" variety. The preparations used in traditional Chinese medicine (TCM), also known as Zhi Shi (枳实), are the immature and dried whole oranges from Citrus aurantium. Extracts of the same material or purified synephrine are also marketed in the US, sometimes in combination with caffeine, as a weight-loss-promoting dietary supplement for oral consumption. While the traditional preparations have been in use for millennia as a component of TCM-formulas, synephrine itself is not an approved over the counter drug. As a pharmaceutical, m-synephrine (phenylephrine) is still used as a sympathomimetic, mostly by injection for the treatment of emergencies such as shock, and rarely orally for the treatment of bronchial problems associated with asthma and hay-fever.

Chlorprothixene, sold under the brand name Truxal among others, is a typical antipsychotic of the thioxanthene group.

D-Lysergic acid α-hydroxyethylamide, also known as D-lysergic acid methyl carbinolamide, is an alkaloid of the ergoline family, believed to be present in various species in the Convolvulaceae, as well as some species of fungi. Morning glory heavenly blue and Hawaiian baby woodrose especially contain high amounts of LSH, with content varying between species and by how fresh.

Hordenine is an alkaloid of the phenethylamine class that occurs naturally in a variety of plants, taking its name from one of the most common, barley. Chemically, hordenine is the N-methyl derivative of N-methyltyramine, and the N,N-dimethyl derivative of the well-known biogenic amine tyramine, from which it is biosynthetically derived and with which it shares some pharmacological properties. As of September 2012, hordenine is widely sold as an ingredient of nutritional supplements, with the claims that it is a stimulant of the central nervous system, and has the ability to promote weight loss by enhancing metabolism. In experimental animals, given sufficiently large doses parenterally, hordenine does produce an increase in blood pressure, as well as other disturbances of the cardiovascular, respiratory, and nervous systems. These effects are generally not reproduced by oral administration of the drug in test animals, and virtually no scientific reports of the effects of hordenine in human beings have been published.

Adrenomedullin is a vasodilator peptide hormone of uncertain significance in human health and disease. It was initially isolated in 1993 from a pheochromocytoma, a tumor of the adrenal medulla: hence the name.
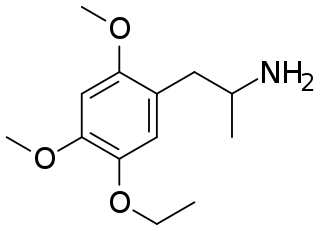
MME (2,4-dimethoxy-5-ethoxyamphetamine) is a lesser-known psychedelic drug. It is a dimethoxy-ethoxy analog of TMA-2. MME was first synthesized by Alexander Shulgin from ethylvanillin. In his book PiHKAL, the minimum dosage is listed as 40 mg and above, and the duration listed as 6–10 hours. Shulgin gives MME a ++ on the Shulgin Rating Scale.

N-Methylphenethylamine (NMPEA) is a naturally occurring trace amine neuromodulator in humans that is derived from the trace amine, phenethylamine (PEA). It has been detected in human urine and is produced by phenylethanolamine N-methyltransferase with phenethylamine as a substrate, which significantly increases PEA's effects. PEA breaks down into Phenylacetaldehyde which is further broken down into Phenylacetic acid by Monoamine oxidase. When this is inhibited by Monoamine oxidase inhibitors, it allows more of the PEA to be metabolized into nymphetamine (NMPEA) and not wasted on the weaker inactive metabolites.

The alpha-2C adrenergic receptor, also known as ADRA2C, is an alpha-2 adrenergic receptor, and also denotes the human gene encoding it.

Deoxyepinephrine, also known by the common names N-methyldopamine and epinine, is an organic compound and natural product that is structurally related to the important neurotransmitters dopamine and epinephrine. All three of these compounds also belong to the catecholamine family. The pharmacology of epinine largely resembles that of its "parent", dopamine. Epinine has been found in plants, insects and animals. It is also of significance as the active metabolic breakdown product of the prodrug ibopamine, which has been used to treat congestive heart failure.

ICI-118,551 is a selective β2 adrenergic receptor (adrenoreceptor) antagonist or beta blocker. ICI binds to the β2 subtype with at least 100 times greater affinity than β1 or β3, the two other known subtypes of the beta adrenoceptor. The compound was developed by Imperial Chemical Industries, which was acquired by AkzoNobel in 2008.

Phenylethanolamine, or β-hydroxyphenethylamine, is a trace amine with a structure similar to those of other trace phenethylamines as well as the catecholamine neurotransmitters dopamine, norepinephrine, and epinephrine. As an organic compound, phenylethanolamine is a β-hydroxylated phenethylamine that is also structurally related to a number of synthetic drugs in the substituted phenethylamine class. In common with these compounds, phenylethanolamine has strong cardiovascular activity and, under the name Apophedrin, has been used as a drug to produce topical vasoconstriction.

A monoamine releasing agent (MRA), or simply monoamine releaser, is a drug that induces the release of a monoamine neurotransmitter from the presynaptic neuron into the synapse, leading to an increase in the extracellular concentrations of the neurotransmitter. Many drugs induce their effects in the body and/or brain via the release of monoamine neurotransmitters, e.g., trace amines, many substituted amphetamines, and related compounds.

4-Substituted-2,5-dimethoxyamphetamines (DOx) is a chemical class of substituted amphetamine derivatives featuring methoxy groups at the 2- and 5- positions of the phenyl ring, and a substituent such as alkyl or halogen at the 4- position of the phenyl ring. Most compounds of this class are potent and long-lasting psychedelic drugs, and act as highly selective 5-HT2A, 5-HT2B, and 5-HT2C receptor partial agonists. A few bulkier derivatives such as DOAM have similarly high binding affinity for 5-HT2 receptors but instead act as antagonists, and so do not produce psychedelic effects though they retain amphetamine-like stimulant effects.

Osemozotan (MKC-242) is a selective 5-HT1A receptor agonist with some functional selectivity, acting as a full agonist at presynaptic and a partial agonist at postsynaptic 5-HT1A receptors. 5-HT1A receptor stimulation influences the release of various neurotransmitters including serotonin, dopamine, norepinephrine, and acetylcholine. 5-HT1A receptors are inhibitory G protein-coupled receptor. Osemozotan has antidepressant, anxiolytic, antiobsessional, serenic, and analgesic effects in animal studies, and is used to investigate the role of 5-HT1A receptors in modulating the release of dopamine and serotonin in the brain, and their involvement in addiction to abused stimulants such as cocaine and methamphetamine.

N-Methyltyramine (NMT), also known as 4-hydroxy-N-methylphenethylamine, is a human trace amine and natural phenethylamine alkaloid found in a variety of plants. As the name implies, it is the N-methyl analog of tyramine, which is a well-known biogenic trace amine with which NMT shares many pharmacological properties. Biosynthetically, NMT is produced by the N-methylation of tyramine via the action of the enzyme phenylethanolamine N-methyltransferase in humans and tyramine N-methyltransferase in plants.

Candicine is a naturally occurring organic compound that is a quaternary ammonium salt with a phenethylamine skeleton. It is the N,N,N-trimethyl derivative of the well-known biogenic amine tyramine, and, being a natural product with a positively charged nitrogen atom in its molecular structure, it is classed as an alkaloid. Although it is found in a variety of plants, including barley, its properties have not been extensively studied with modern techniques. Candicine is toxic after parenteral administration, producing symptoms of neuromuscular blockade; further details are given in the "Pharmacology" section below.

Halostachine is a natural product, an alkaloid first isolated from the Asian shrub Halostachys caspica, and structurally a β-hydroxy-phenethylamine related to its better-known "parent" biogenic amine, phenylethanolamine, to the adrenergic drug synephrine, and to the alkaloid ephedrine. The pharmacological properties of halostachine have some similarity to those of these structurally-related compounds, and Halostachys caspica extracts have been included as a constituent of certain OTC dietary supplements, but halostachine has never been developed as a prescription drug. Although it is found in nature as a single stereoisomer, halostachine is more commonly available as a synthetic product in the form of its racemate. In appearance it is a colorless solid.
