
Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders. Symptoms often include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis. The current classification was last updated in 2017, when a number of rarer forms of EDS were added.

Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin. The degree to which a person is affected, however, may vary from mild to severe. Complications may include breathing problems, problems seeing, cleft palate, and hearing loss. Those affected generally have normal intelligence.

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.

Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis. This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly). Individuals with more severe cases of SCS may have mild to moderate intellectual or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention. Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.

Fraser syndrome is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.

Simpson–Golabi–Behmel syndrome (SGBS), is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.

Pallister–Hall syndrome (PHS) is a rare genetic disorder that affects various body systems. The main features are a non-cancerous mass on the hypothalamus and extra digits (polydactylism). The prevalence of Pallister-Hall Syndrome is unknown; about 100 cases have been reported in publication.
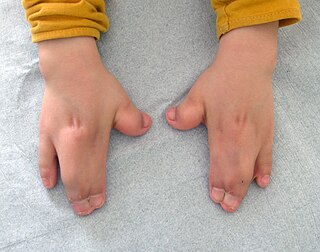
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.

Twist-related protein 1 (TWIST1) also known as class A basic helix–loop–helix protein 38 (bHLHa38) is a basic helix-loop-helix transcription factor that in humans is encoded by the TWIST1 gene.
Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.

Twist-related protein 2 is a protein that in humans is encoded by the TWIST2 gene. The protein encoded by this gene is a basic helix-loop-helix (bHLH) transcription factor and shares similarity with another bHLH transcription factor, TWIST1. bHLH transcription factors have been implicated in cell lineage determination and differentiation. It is thought that during osteoblast development, this protein may inhibit osteoblast maturation and maintain cells in a preosteoblast phenotype.

Frank–Ter Haar syndrome (FTHS), also known as Ter Haar-syndrome, is a rare disease characterized by abnormalities that affect bone, heart, and eye development. Children born with the disease usually die very young.

Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.

Macrostomia refers to a mouth that is unusually wide. The term is from the Greek prefix makro- meaning "large" and from Greek στόμα, "mouth".

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
A facial cleft is an opening or gap in the face, or a malformation of a part of the face. Facial clefts is a collective term for all sorts of clefts. All structures like bone, soft tissue, skin etc. can be affected. Facial clefts are extremely rare congenital anomalies. There are many variations of a type of clefting and classifications are needed to describe and classify all types of clefting. Facial clefts hardly ever occur isolated; most of the time there is an overlap of adjacent facial clefts.
Nasodigitoacoustic syndrome, also called Keipert syndrome, is a rare congenital syndrome first described by J.A. Keipert and colleagues in 1973. The syndrome is characterized by a misshaped nose, broad thumbs and halluces, brachydactyly, sensorineural hearing loss, facial features such as hypertelorism, and developmental delay.
Barber-Say syndrome (BSS) is a very rare congenital disorder associated with excessive hair growth (hypertrichosis), fragile (atrophic) skin, eyelid deformities (ectropion), and an overly broad mouth (macrostomia).
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.
