Myelin is a lipid-rich material that surrounds nerve cell axons to insulate them and increase the rate at which electrical impulses pass along the axon. The myelinated axon can be likened to an electrical wire with insulating material (myelin) around it. However, unlike the plastic covering on an electrical wire, myelin does not form a single long sheath over the entire length of the axon. Rather, myelin ensheaths the axon segmentally: in general, each axon is encased in multiple long sheaths with short gaps between, called nodes of Ranvier. At the nodes of Ranvier, which are approximately one thousandth of a mm in length, the axon's membrane is bare of myelin.
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.
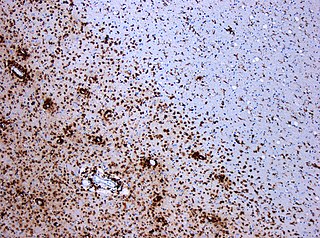
A demyelinating disease refers to any disease affecting the nervous system where the myelin sheath surrounding neurons is damaged. This damage disrupts the transmission of signals through the affected nerves, resulting in a decrease in their conduction ability. Consequently, this reduction in conduction can lead to deficiencies in sensation, movement, cognition, or other functions depending on the nerves affected.

Alexander disease is a very rare autosomal dominant leukodystrophy, which are neurological conditions caused by anomalies in the myelin which protects nerve fibers in the brain. The most common type is the infantile form that usually begins during the first two years of life. Symptoms include mental and physical developmental delays, followed by the loss of developmental milestones, an abnormal increase in head size and seizures. The juvenile form of Alexander disease has an onset between the ages of 2 and 13 years. These children may have excessive vomiting, difficulty swallowing and speaking, poor coordination, and loss of motor control. Adult-onset forms of Alexander disease are less common. The symptoms sometimes mimic those of Parkinson’s disease or multiple sclerosis, or may present primarily as a psychiatric disorder.

CADASIL or CADASIL syndrome, involving cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, is the most common form of hereditary stroke disorder, and is thought to be caused by mutations of the Notch 3 gene on chromosome 19. The disease belongs to a family of disorders called the leukodystrophies. The most common clinical manifestations are migraine headaches and transient ischemic attacks or strokes, which usually occur between 40 and 50 years of age, although MRI is able to detect signs of the disease years prior to clinical manifestation of disease.

Krabbe disease (KD) is a rare and often fatal lysosomal storage disease that results in progressive damage to the nervous system. KD involves dysfunctional metabolism of sphingolipids and is inherited in an autosomal recessive pattern. The disease is named after the Danish neurologist Knud Krabbe (1885–1961).

Leukodystrophies are a group of, usually, inherited disorders, characterized by degeneration of the white matter in the brain. The word leukodystrophy comes from the Greek roots leuko, "white", dys, "abnormal" and troph, "growth". The leukodystrophies are caused by imperfect growth or development of the glial cells which produce the myelin sheath, the fatty insulating covering around nerve fibers. Leukodystrophies may be classified as hypomyelinating or demyelinating diseases, respectively, depending on whether the damage is present before birth or occurs after. Other demyelinating diseases are usually not congenital and have a toxic or autoimmune cause.
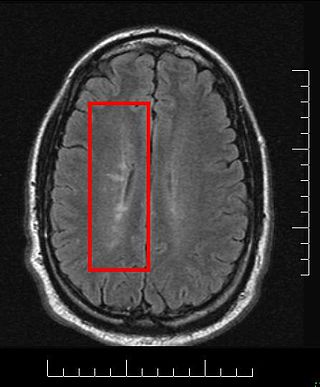
Multiple sclerosis and other demyelinating diseases of the central nervous system (CNS) produce lesions and glial scars or scleroses. They present different shapes and histological findings according to the underlying condition that produces them.
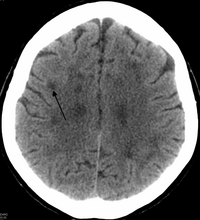
Leukoencephalopathy is a term that describes all of the brain white matter diseases, whether their molecular cause is known or unknown. It can refer specifically to any of these diseases:

Leukoencephalopathy with vanishing white matter is an autosomal recessive neurological disease. The cause of the disease are mutations in any of the 5 genes encoding subunits of the translation initiation factor eIF2B: EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5. The disease belongs to a family of conditions called the Leukodystrophies.
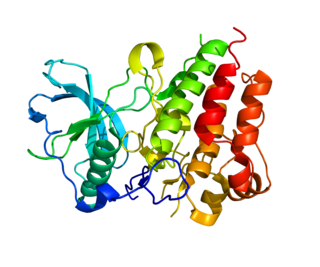
Colony stimulating factor 1 receptor (CSF1R), also known as macrophage colony-stimulating factor receptor (M-CSFR), and CD115, is a cell-surface protein encoded by the human CSF1R gene. CSF1R is a receptor that can be activated by two ligands: colony stimulating factor 1 (CSF-1) and interleukin-34 (IL-34). CSF1R is highly expressed in myeloid cells, and CSF1R signaling is necessary for the survival, proliferation, and differentiation of many myeloid cell types in vivo and in vitro. CSF1R signaling is involved in many diseases and is targeted in therapies for cancer, neurodegeneration, and inflammatory bone diseases.
Leukoencephalopathy with neuroaxonal spheroids (LENAS) is an extremely rare kind of leukoencephalopathy and is classified as a neurodegenerative disease. LENAS is a cause of severe and subacute dementia that results from damage to certain areas of the brain. This damage is to a type of brain tissue called white matter and axon damage due to swellings which are termed spheroids.
A hyperintensity or T2 hyperintensity is an area of high intensity on types of magnetic resonance imaging (MRI) scans of the brain of a human or of another mammal that reflect lesions produced largely by demyelination and axonal loss. These small regions of high intensity are observed on T2 weighted MRI images within cerebral white matter or subcortical gray matter. The volume and frequency is strongly associated with increasing age. They are also seen in a number of neurological disorders and psychiatric illnesses. For example, deep white matter hyperintensities are 2.5 to 3 times more likely to occur in bipolar disorder and major depressive disorder than control subjects. WMH volume, calculated as a potential diagnostic measure, has been shown to correlate to certain cognitive factors. Hyperintensities appear as "bright signals" on an MRI image and the term "bright signal" is occasionally used as a synonym for a hyperintensity.
Megalencephalic leukoencephalopathy with subcortical cysts is a form of hereditary CNS demyelinating disease. It belongs to a group of disorders called leukodystrophies. It is characterized by early-onset enlargement of the head (macrocephaly) as well as delayed-onset neurological deterioration to include spasticity, epilepsy, and lack of muscular coordination. MLC does not appear to be a disease that is fatal at birth or early in life despite its symptoms, although the number of patients throughout history known to have the disease is fairly limited.
Grinker's myelinopathy, also known as anoxic leukoencephalopathy, is a rare disease of the central nervous system. The disease is characterized by a delayed leukoencephalopathy after a hypoxic episode. It is typically, though not necessarily, related to carbon monoxide poisoning or heroin overdose. It occurs in roughly 2.8% of those who experience an acute hypoxic/anoxic episode. Because of the wide range of symptoms and the delay in onset, it is often misdiagnosed as other neuropathologies. Grinker's myelinopathy was originally characterized by Roy R. Grinker in 1925 or 1926, depending on the source.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is disease of the arteries in the brain, which causes tissue loss in the subcortical region of the brain and the destruction of myelin in the CNS. CARASIL is characterized by symptoms such as gait disturbances, hair loss, low back pain, dementia, and stroke. CARASIL is a rare disease, having only been diagnosed in about 50 patients, of which ten have been genetically confirmed. Most cases have been reported in Japan, but Chinese and caucasian individuals have also been diagnosed with the disease. CARASIL is inherited in an autosomal recessive pattern. There is currently no cure for CARASIL. Other names for CARASIL include familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension, Nemoto disease and Maeda syndrome.
Nasu–Hakola disease also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy is a rare disease characterised by early-onset dementia and multifocal bone cysts. It is caused by autosomal recessive loss of function mutations in either the TREM2 or TYROBP gene that are found most frequently in the Finnish and Japanese populations.
Hypomyelination-congenital cataract syndrome is a rare autosomal recessive hereditary disorder that affects the brain's white matter and is characterized by congenital cataract, psychomotor development delays, and moderate intellectual disabilities. It is a type of leukoencephalopathy.

Autosomal dominant leukodystrophy with autonomic disease is a rare neurological condition of genetic origin which is characterized by gradual demyelination of the central nervous system which results in various impairments, including ataxia, mild cognitive disability and autonomic dysfunction. It is part of a group of disorders called "leukodystrophies".