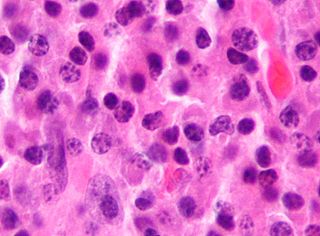
Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making aplasia, myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.
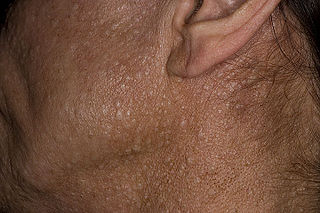
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons is a human, adult onset, autosomal dominant genetic disorder caused by the FLCN gene. It can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%) and 24% of people with BHD eventually experience a collapsed lung. Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.

Langerhans cell histiocytosis (LCH) is an abnormal clonal proliferation of Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes.

Sturge–Weber syndrome, sometimes referred to as encephalotrigeminal angiomatosis, is a rare congenital neurological and skin disorder. It is one of the phakomatoses and is often associated with port-wine stains of the face, glaucoma, seizures, intellectual disability, and ipsilateral leptomeningeal angioma. Sturge–Weber syndrome can be classified into three different types. Type 1 includes facial and leptomeningeal angiomas as well as the possibility of glaucoma or choroidal lesions. Normally, only one side of the brain is affected. This type is the most common. Type 2 involvement includes a facial angioma with a possibility of glaucoma developing. There is no evidence of brain involvement. Symptoms can show at any time beyond the initial diagnosis of the facial angioma. The symptoms can include glaucoma, cerebral blood flow abnormalities and headaches. More research is needed on this type of Sturge–Weber syndrome. Type 3 has leptomeningeal angioma involvement exclusively. The facial angioma is absent and glaucoma rarely occurs. This type is only diagnosed via brain scan.

Hyphema is the medical condition of bleeding in the anterior chamber of the eye between the iris and the cornea. People usually first notice a loss or decrease in vision. The eye may also appear to have a reddish tinge, or it may appear as a small pool of blood at the bottom of the iris in the cornea. A traumatic hyphema is caused by a blow to the eye. A hyphema can also occur spontaneously.

Letterer–Siwe disease, (LSD) or Abt-Letterer-Siwe disease, is one of the four recognized clinical syndromes of Langerhans cell histiocytosis (LCH) and is the most severe form, involving multiple organ systems such as the skin, bone marrow, spleen, liver, and lung. Oral cavity and gastrointestinal involvement may also be seen. LCH and all its subtypes are characterized by monoclonal migration and proliferation of specific dendritic cells.
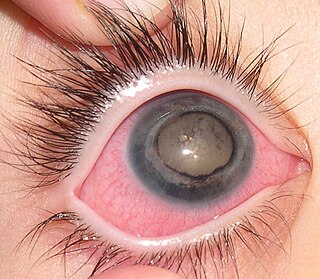
Coats' disease is a rare congenital, nonhereditary eye disorder, causing full or partial blindness, characterized by abnormal development of blood vessels behind the retina. Coats' disease can also fall under glaucoma.

A histiocytoma in the dog is a benign tumor. It is an abnormal growth in the skin of histiocytes (histiocytosis), a cell that is part of the immune system. A similar disease in humans, Hashimoto-Pritzker disease, is also a Langerhans cell histiocytosis. Dog breeds that may be more at risk for this tumor include Bulldogs, American Pit Bull Terriers, American Staffordshire Terriers, Scottish Terriers, Greyhounds, Boxers, and Boston Terriers. They also rarely occur in goats and cattle.
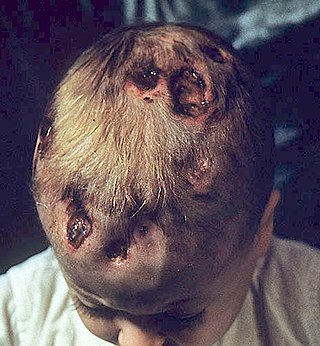
Chronic multifocal Langerhans cell histiocytosis, previously known as Hand–Schüller–Christian disease, is a type of Langerhans cell histiocytosis (LCH), which can affect multiple organs. The condition is traditionally associated with a combination of three features; bulging eyes, breakdown of bone, and diabetes insipidus, although around 75% of cases do not have all three features. Other features may include a fever and weight loss, and depending on the organs involved there may be rashes, asymmetry of the face, ear infections, signs in the mouth and the appearance of advanced gum disease. Features relating to lung and liver disease may occur.

Rosai–Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy or sometimes as Destombes–Rosai–Dorfman disease, is a rare disorder of unknown cause that is characterized by abundant histiocytes in the lymph nodes or other locations throughout the body.
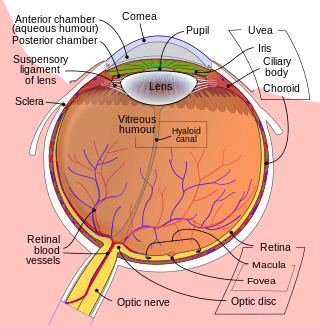
Intraocular hemorrhage is bleeding inside the eye. Bleeding can occur from any structure of the eye where there is vasculature or blood flow, including the anterior chamber, vitreous cavity, retina, choroid, suprachoroidal space, or optic disc.

Blueberry muffin baby, also known as extramedullary hematopoiesis, describes a newborn baby with multiple purpura, associated with several non-cancerous and cancerous conditions in which extra blood is produced in the skin. The bumps range from one to seven mm, do not blanch and have a tendency to occur on the head, neck and trunk. They often fade by three to six weeks after birth, leaving brownish marks. When due to a cancer, the bumps tend to be fewer, firmer and larger.

Langerhans cell sarcoma (LCS) is a rare form of malignant histiocytosis. It should not be confused with Langerhans cell histiocytosis, which is cytologically benign. It can present most commonly in the skin and lymphatic tissue, but may also present in the lung, liver, and bone marrow. Treatment is most commonly with surgery or chemotherapy.
Non-X histiocytoses are a clinically well-defined group of cutaneous syndromes characterized by infiltrates of monocytes/macrophages, as opposed to X-type histiocytoses in which the infiltrates contain Langerhans cells. Conditions included in this group are:
Indeterminate cell histiocytosis(LCH) is an uncommon proliferative illness where the predominant cells have characteristics from both non-Langerhans cell histiocytosis (NLCH) and Langerhans cell histiocytosis (LCH) in terms of morphology and immunophenotypic characteristics. Wood et al. originally described ICH in 1985 as a neoplastic disease arising from dermal indeterminate cells that lack Birbeck granules but are characteristically positive for S-100 and CD1a.
Congenital self-healing reticulohistiocytosis is a condition that is a self-limited form of Langerhans cell histiocytosis.

Bonnet–Dechaume–Blanc syndrome, also known as Wyburn-Mason syndrome, is a rare congenital disorder characterized by arteriovenous malformations of the brain, retina or facial nevi. The syndrome has a number of possible symptoms and can, more rarely, affect the skin, bones, kidneys, muscles, and gastrointestinal tract. When the syndrome affects the brain, people can experience severe headaches, seizures, acute stroke, meningism, and progressive neurological deficits due to acute or chronic ischaemia caused by arteriovenous shunting.
Histiocytic diseases in dogs are a group of diseases in dogs which may involve the skin, and which can be difficult to differentiate from granulomatous, reactive inflammatory or lymphoproliferative diseases. The clinical presentation and behaviour as well as response to therapy vary greatly among the syndromes.
Crystal-storing histiocytosis is a form of histiocytosis which mostly occurs in people with monoclonal gammopathies. Histiocytosis is an excessive number of histiocytes. In the vast majority of crystal-storing histiocytosis cases, immunoglobulins accumulate within the cytoplasm of histiocytes; in rare cases clofazimine, cystine, silica, or Charcot–Leyden crystals may be found in the histiocytes instead. Non-immunoglobulin crystal-storing histiocytosis is mostly associated with non-malignant disorders, such as chronic inflammation or autoimmune abnormality conditions such as rheumatoid arthritis, Crohn's disease, or Helicobacter pylori gastritis. It may be a localised or generalised disease. Examples of locations where histiocytosis may occur include the lungs, pleura, stomach, kidney, bone marrow, thyroid, thymus, and parotid gland. The disease is described as generalised if two or more unrelated sites are involved.

