Signs and symptoms
| | This section is empty. You can help by adding to it. (November 2023) |
| Permanent neonatal diabetes mellitus | |
|---|---|
| Other names | PNDM |
| Specialty | Neonatology |
Permanent neonatal diabetes mellitus (PNDM) is a newly identified and potentially treatable form of monogenic diabetes. This type of neonatal diabetes is caused by activating mutations of the KCNJ11 gene, which codes for the Kir6.2 subunit of the beta cell KATP channel. [1] [2] This disease is considered to be a type of maturity onset diabetes of the young (MODY).
| | This section is empty. You can help by adding to it. (November 2023) |
It can be associated with GCK , KCNJ11 , INS , and ABCC8 . [3]
This results in congenital impairment of insulin release, although in the past, this was always being thought to be unusually early type 1 diabetes mellitus. The insulin deficiency results in intrauterine growth retardation with birth weight small for gestational age. The diabetes is usually diagnosed in the first 3 months of life due to continuing poor weight gain, polyuria, or diabetic ketoacidosis. Rare cases have been recognized as late as 6 months of age.
Remarkably, this type of diabetes often responds well to sulfonylureas and insulin may not be necessary. More severe mutations in the KCNJ11 gene can cause early-onset diabetes which does not respond to the sulfonylurea drugs, as well as a syndrome of developmental delay and neurological features called the DEND syndrome. These forms of diabetes are very rare conditions, appearing in about 1/100,000 to 1/200,000 live births, and accounting for about 1/1000 of type 1 diabetes cases. Fewer than 5% of the cases assumed to exist have been diagnosed, and most diabetes clinics around the world are checking for KCNJ11 mutations in any persons who developed apparent insulin-dependent diabetes without the typical type 1 antibodies before 6 months of age. At least some of these people have been able to change from insulin to sulfonylurea pills after decades of injections.[ citation needed ]

Wolfram syndrome, also called DIDMOAD, is a rare autosomal-recessive genetic disorder that causes childhood-onset diabetes mellitus, optic atrophy, and deafness as well as various other possible disorders including neurodegeneration.
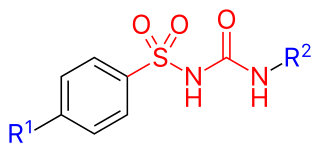
Sulfonylureas or sulphonylureas are a class of organic compounds used in medicine and agriculture. The functional group consists of a sulfonyl group (-S(=O)2) with its sulphur atom bonded to a nitrogen atom of a ureylene group (N,N-dehydrourea, a dehydrogenated derivative of urea). The side chains R1 and R2 distinguish various sulfonylureas.
Maturity-onset diabetes of the young (MODY) refers to any of several hereditary forms of diabetes mellitus caused by mutations in an autosomal dominant gene disrupting insulin production. Along with neonatal diabetes, MODY is a form of the conditions known as monogenic diabetes. While the more common types of diabetes involve more complex combinations of causes involving multiple genes and environmental factors, each forms of MODY are caused by changes to a single gene (monogenic). GCK-MODY and HNF1A-MODY are the most common forms.

Congenital hyperinsulinism (HI or CHI) is a rare condition causing severe hypoglycemia in newborns due to the overproduction of insulin. There are various causes of HI, some of which are known to be the result of a genetic mutation. Sometimes HI occurs on its own (isolated) and more rarely associated with other medical conditions.

For pregnant women with diabetes, some particular challenges exist for both mother and fetus. If the pregnant woman has diabetes as a pre-existing disorder, it can cause early labor, birth defects, and larger than average infants. Therefore, experts advise diabetics to maintain blood sugar level close to normal range about 3 months before planning for pregnancy.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
An ATP-sensitive potassium channel is a type of potassium channel that is gated by intracellular nucleotides, ATP and ADP. ATP-sensitive potassium channels are composed of Kir6.x-type subunits and sulfonylurea receptor (SUR) subunits, along with additional components. KATP channels are found in the plasma membrane; however some may also be found on subcellular membranes. These latter classes of KATP channels can be classified as being either sarcolemmal ("sarcKATP"), mitochondrial ("mitoKATP"), or nuclear ("nucKATP").

Kir6.2 is a major subunit of the ATP-sensitive K+ channel, a lipid-gated inward-rectifier potassium ion channel. The gene encoding the channel is called KCNJ11 and mutations in this gene are associated with congenital hyperinsulinism.
Hepatocyte nuclear factor 4 alpha (HNF4A) also known as NR2A1 is a nuclear receptor that in humans is encoded by the HNF4A gene.

HNF1 homeobox A, also known as HNF1A, is a human gene on chromosome 12. It is ubiquitously expressed in many tissues and cell types. The protein encoded by this gene is a transcription factor that is highly expressed in the liver and is involved in the regulation of the expression of several liver-specific genes. Mutations in the HNF1A gene have been known to cause diabetes. The HNF1A gene also contains a SNP associated with increased risk of coronary artery disease.
ATP-binding cassette transporter sub-family C member 8 is a protein that in humans is encoded by the ABCC8 gene. ABCC8 orthologs have been identified in all mammals for which complete genome data are available.

Wolcott–Rallison syndrome,WRS, is a rare, autosomal recessive disorder with infancy-onset diabetes mellitus, multiple epiphyseal dysplasia, osteopenia, mental retardation or developmental delay, and hepatic and renal dysfunction as main clinical findings. Patients with WRS have mutations in the EIF2AK3 gene, which encodes the eukaryotic translation initiation factor 2-alpha kinase 3. Other disease names include multiple epiphyseal dysplasia and early-onset diabetes mellitus. Most patients with this disease do not survive to adulthood. The majority of WRS patients die from fulminant hepatitis during childhood. There are few reported cases for this disease. Of the 54 families worldwide with reported WRS cases, 22.2% of them are from the Kingdom of Saudi Arabia. Of the 23 WRS patients in Saudi Arabia, all but one is the result of consanguineous marriages. Another country where WRS cases have been found is Kosovo. Here, the Albanian population is also known for consanguineous marriages, but there were some cases involving patients from non-consanguineous parents that were carriers for the same mutant allele.
MODY 2 or GCK-MODY is a form of maturity-onset diabetes of the young. It is due to any of several mutations in the GCK gene on human chromosome 7 for glucokinase. Glucokinase serves as the glucose sensor for the pancreatic beta cell. Normal glucokinase triggers insulin secretion as the glucose exceeds about 90 mg/dl. These loss-of-function mutations result in a glucokinase molecule that is less sensitive or less responsive to rising levels of glucose. The beta cells in MODY 2 have a normal ability to make and secrete insulin, but do so only above an abnormally high threshold. This produces a chronic, mild increase in blood sugar, which is usually asymptomatic. It is usually detected by accidental discovery of mildly elevated blood sugar. An oral glucose tolerance test is much less abnormal than would be expected from the impaired (elevated) fasting blood sugar, since insulin secretion is usually normal once the glucose has exceeded the threshold for that specific variant of the glucokinase enzyme.
MODY 3 or HNF1A-MODY is a form of maturity-onset diabetes of the young. It is caused by mutations of the HNF1-alpha gene, a homeobox gene on human chromosome 12. This is the most common type of MODY in populations with European ancestry, accounting for about 70% of all cases in Europe. HNF1α is a transcription factor that is thought to control a regulatory network important for differentiation of beta cells. Mutations of this gene lead to reduced beta cell mass or impaired function. MODY 1 and MODY 3 diabetes are clinically similar. About 70% of people develop this type of diabetes by age 25 years, but it occurs at much later ages in a few. This type of diabetes can often be treated with sulfonylureas with excellent results for decades. However, the loss of insulin secretory capacity is slowly progressive and most eventually need insulin.
MODY 4 or PDX1-MODY is a form of maturity onset diabetes of the young.

Renal cysts and diabetes syndrome (RCAD), also known as MODY 5 or HNF1B-MODY, is a form of maturity onset diabetes of the young.
Transient neonatal diabetes mellitus (TNDM) is a form of neonatal diabetes presenting at birth that is not permanent. This disease is considered to be a type of maturity onset diabetes of the young (MODY).

Neonatal diabetes mellitus (NDM) is a disease that affects an infant and their body's ability to produce or use insulin.NDM is a kind of diabetes that is monogenic and arises in the first 6 months of life. Infants do not produce enough insulin, leading to an increase in glucose accumulation. It is a rare disease, occurring in only one in 100,000 to 500,000 live births. NDM can be mistaken for the much more common type 1 diabetes, but type 1 diabetes usually occurs later than the first 6 months of life. There are two types of NDM: permanent neonatal diabetes mellitus (PNDM), a lifelong condition, and transient neonatal diabetes mellitus (TNDM), a form of diabetes that disappears during the infant stage but may reappear later in life.
MODY 6 or NEUROD1-MODY is a form of maturity onset diabetes of the young.
Most cases of type 2 diabetes involved many genes contributing small amount to the overall condition. As of 2011 more than 36 genes have been found that contribute to the risk of type 2 diabetes. All of these genes together still only account for 10% of the total genetic component of the disease.