
The β-sheet is a common motif of regular secondary structure in proteins. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in formation of the protein aggregates and fibrils observed in many human diseases, notably the amyloidoses such as Alzheimer's disease.
DnaG is a bacterial DNA primase and is encoded by the dnaG gene. The enzyme DnaG, and any other DNA primase, synthesizes short strands of RNA known as oligonucleotides during DNA replication. These oligonucleotides are known as primers because they act as a starting point for DNA synthesis. DnaG catalyzes the synthesis of oligonucleotides that are 10 to 60 nucleotides long, however most of the oligonucleotides synthesized are 11 nucleotides. These RNA oligonucleotides serve as primers, or starting points, for DNA synthesis by bacterial DNA polymerase III. DnaG is important in bacterial DNA replication because DNA polymerase cannot initiate the synthesis of a DNA strand, but can only add nucleotides to a preexisting strand. DnaG synthesizes a single RNA primer at the origin of replication. This primer serves to prime leading strand DNA synthesis. For the other parental strand, the lagging strand, DnaG synthesizes an RNA primer every few kilobases (kb). These primers serve as substrates for the synthesis of Okazaki fragments.

Histone acetyltransferases (HATs) are enzymes that acetylate conserved lysine amino acids on histone proteins by transferring an acetyl group from acetyl-CoA to form ε-N-acetyllysine. DNA is wrapped around histones, and, by transferring an acetyl group to the histones, genes can be turned on and off. In general, histone acetylation increases gene expression.
In biology and biochemistry, protease inhibitors, or antiproteases, are molecules that inhibit the function of proteases. Many naturally occurring protease inhibitors are proteins.
A supersecondary structure is a compact three-dimensional protein structure of several adjacent elements of a secondary structure that is smaller than a protein domain or a subunit. Supersecondary structures can act as nucleations in the process of protein folding.

T-box transcription factor TBX1 also known as T-box protein 1 and testis-specific T-box protein is a protein that in humans is encoded by the TBX1 gene. Genes in the T-box family are transcription factors that play important roles in the formation of tissues and organs during embryonic development. To carry out these roles, proteins made by this gene family bind to specific areas of DNA called T-box binding element (TBE) to control the expression of target genes.

The TIM barrel, also known as an α/β barrel, is a conserved protein fold consisting of eight α-helices and eight parallel β-strands that alternate along the peptide backbone. The structure is named after triosephosphate isomerase, a conserved metabolic enzyme. TIM barrels are ubiquitous, with approximately 10% of all enzymes adopting this fold. Further, 5 of 7 enzyme commission (EC) enzyme classes include TIM barrel proteins. The TIM barrel fold is evolutionarily ancient, with many of its members possessing little similarity today, instead falling within the twilight zone of sequence similarity.

Gamma-aminobutyric acid receptor subunit beta-3 is a protein that in humans is encoded by the GABRB3 gene. It is located within the 15q12 region in the human genome and spans 250kb. This gene includes 10 exons within its coding region. Due to alternative splicing, the gene codes for many protein isoforms, all being subunits in the GABAA receptor, a ligand-gated ion channel. The beta-3 subunit is expressed at different levels within the cerebral cortex, hippocampus, cerebellum, thalamus, olivary body and piriform cortex of the brain at different points of development and maturity. GABRB3 deficiencies are implicated in many human neurodevelopmental disorders and syndromes such as Angelman syndrome, Prader-Willi syndrome, nonsyndromic orofacial clefts, epilepsy and autism. The effects of methaqualone and etomidate are mediated through GABBR3 positive allosteric modulation.

Histidine kinases (HK) are multifunctional, and in non-animal kingdoms, typically transmembrane, proteins of the transferase class of enzymes that play a role in signal transduction across the cellular membrane. The vast majority of HKs are homodimers that exhibit autokinase, phosphotransfer, and phosphatase activity. HKs can act as cellular receptors for signaling molecules in a way analogous to tyrosine kinase receptors (RTK). Multifunctional receptor molecules such as HKs and RTKs typically have portions on the outside of the cell that bind to hormone- or growth factor-like molecules, portions that span the cell membrane, and portions within the cell that contain the enzymatic activity. In addition to kinase activity, the intracellular domains typically have regions that bind to a secondary effector molecule or complex of molecules that further propagate signal transduction within the cell. Distinct from other classes of protein kinases, HKs are usually parts of a two-component signal transduction mechanisms in which HK transfers a phosphate group from ATP to a histidine residue within the kinase, and then to an aspartate residue on the receiver domain of a response regulator protein. More recently, the widespread existence of protein histidine phosphorylation distinct from that of two-component histidine kinases has been recognised in human cells. In marked contrast to Ser, Thr and Tyr phosphorylation, the analysis of phosphorylated Histidine using standard biochemical and mass spectrometric approaches is much more challenging, and special procedures and separation techniques are required for their preservation alongside classical Ser, Thr and Tyr phosphorylation on proteins isolated from human cells.

Long-chain-fatty-acid—CoA ligase 4 is an enzyme that in humans is encoded by the ACSL4 gene.
T-box transcription factor TBX22 is a protein that in humans is encoded by the TBX22 gene.
X-linked intellectual disability refers to forms of intellectual disability which are specifically associated with X-linked recessive inheritance.
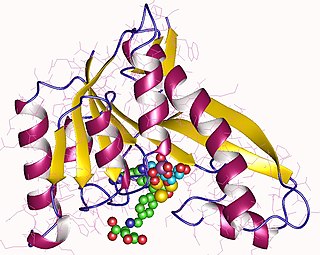
Phosphoribosylglycinamide formyltransferase (EC 2.1.2.2, 2-amino-N-ribosylacetamide 5'-phosphate transformylase, GAR formyltransferase, GAR transformylase, glycinamide ribonucleotide transformylase, GAR TFase, 5,10-methenyltetrahydrofolate:2-amino-N-ribosylacetamide ribonucleotide transformylase) is an enzyme with systematic name 10-formyltetrahydrofolate:5'-phosphoribosylglycinamide N-formyltransferase. This enzyme catalyses the following chemical reaction
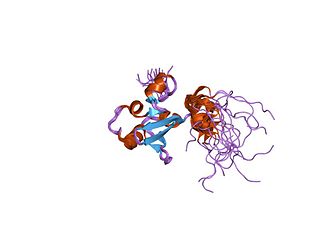
In molecular biology, the protein domain SAND is named after a range of proteins in the protein family: Sp100, AIRE-1, NucP41/75, DEAF-1. It is localised in the cell nucleus and has an important function in chromatin-dependent transcriptional control. It is found solely in eukaryotes.

In molecular biology, a carbohydrate-binding module (CBM) is a protein domain found in carbohydrate-active enzymes. The majority of these domains have carbohydrate-binding activity. Some of these domains are found on cellulosomal scaffoldin proteins. CBMs were previously known as cellulose-binding domains. CBMs are classified into numerous families, based on amino acid sequence similarity. There are currently 64 families of CBM in the CAZy database.

ClpS is an N-recognin in the N-end rule pathway. ClpS interacts with protein substrates that have a bulky hydrophobic residue at the N-terminus. The protein substrate is then degraded by the ClpAP protease.

Coiled-coil domain-containing protein 144A is a protein that in humans is encoded by the CCDC144A gene. An alias of this gene is called KIAA0565. There are four members of the CCDC family: CCDC 144A, 144B, 144C and putative CCDC 144 N-terminal like proteins.

Transmembrane protein 8A is a protein that in humans is encoded by the TMEM8A gene (16p13.3.). Evolutionarily, TMEM8A orthologs are found in primates and mammals and in a few more distantly related species. TMEM8A contains five transmembrane domains and one EGF-like domain which are all highly conserved in the ortholog space. Although there is no confirmed function of TMEM8A, through analyzing expression and experimental data, it is predicted that TMEM8A is an adhesion protein that plays a role in keeping T-cells in their resting state.
ZC3H12B, also known as CXorf32 or MCPIP2, is a protein encoded by gene ZC3H12B located on chromosome Xq12 in humans.

Biliverdin reductase B is a protein that in humans is encoded by the BLVRB gene.
