A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

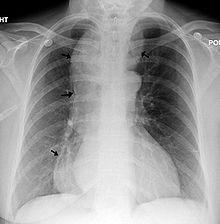
Esophageal achalasia, often referred to simply as achalasia, is a failure of smooth muscle fibers to relax, which can cause the lower esophageal sphincter to remain closed. Without a modifier, "achalasia" usually refers to achalasia of the esophagus. Achalasia can happen at various points along the gastrointestinal tract; achalasia of the rectum, for instance, may occur in Hirschsprung's disease. The lower esophageal sphincter is a muscle between the esophagus and stomach that opens when food comes in. It closes to avoid stomach acids from coming back up. A fully understood cause to the disease is unknown, as are factors that increase the risk of its appearance. Suggestions of a genetically transmittable form of achalasia exist, but this is neither fully understood, nor agreed upon.

Microcephaly is a medical condition involving a smaller-than-normal head. Microcephaly may be present at birth or it may develop in the first few years of life. Brain development is often affected; people with this disorder often have an intellectual disability, poor motor function, poor speech, abnormal facial features, seizures and dwarfism.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Caroli disease is a rare inherited disorder characterized by cystic dilatation of the bile ducts within the liver. There are two patterns of Caroli disease: focal or simple Caroli disease consists of abnormally widened bile ducts affecting an isolated portion of liver. The second form is more diffuse, and when associated with portal hypertension and congenital hepatic fibrosis, is often referred to as "Caroli syndrome". The underlying differences between the two types are not well understood. Caroli disease is also associated with liver failure and polycystic kidney disease. The disease affects about one in 1,000,000 people, with more reported cases of Caroli syndrome than of Caroli disease.

Ichthyosis is a family of genetic skin disorders characterized by dry, thickened, scaly skin. The more than 20 types of ichthyosis range in severity of symptoms, outward appearance, underlying genetic cause and mode of inheritance. Ichthyosis comes from the Greek ἰχθύςichthys, literally 'fish', since dry, scaly skin is the defining feature of all forms of ichthyosis.
Miller–Dieker syndrome, Miller–Dieker lissencephaly syndrome (MDLS), and chromosome 17p13.3 deletion syndrome is a micro deletion syndrome characterized by congenital malformations. Congenital malformations are physical defects detectable in an infant at birth which can involve many different parts of the body including the brain, hearts, lungs, liver, bones, or intestinal tract. MDS is a contiguous gene syndrome – a disorder due to the deletion of multiple gene loci adjacent to one another. The disorder arises from the deletion of part of the small arm of chromosome 17p, leading to partial monosomy. There may be unbalanced translocations, or the presence of a ring chromosome 17.
Malouf syndrome is a congenital disorder that causes one or more of the following symptoms: mental retardation, ovarian dysgenesis, congestive cardiomyopathy, broad nasal base, blepharoptosis, and bone abnormalities, and occasionally marfanoid habitus.

Triple-A syndrome or AAA syndrome is a rare autosomal recessive congenital disorder. In most cases, there is no family history of AAA syndrome. The syndrome was first identified by Jeremy Allgrove and colleagues in 1978; since then just over 100 cases have been reported. The syndrome is called Triple-A due to the manifestation of the illness which includes achalasia, addisonianism, and alacrima. Alacrima is usually the earliest manifestation. Neurodegeneration or atrophy of the nerve cells and autonomic dysfunction may be seen in the disorder; therefore, some have suggested the disorder be called 4A syndrome. It is a progressive disorder that can take years to develop the full-blown clinical picture. The disorder also has variability and heterogeneity in presentation.
Mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE) is a rare autosomal recessive mitochondrial disease. It has been previously referred to as polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudoobstruction. The disease presents in childhood, but often goes unnoticed for decades. Unlike typical mitochondrial diseases caused by mitochondrial DNA (mtDNA) mutations, MNGIE is caused by mutations in the TYMP gene, which encodes the enzyme thymidine phosphorylase. Mutations in this gene result in impaired mitochondrial function, leading to intestinal symptoms as well as neuro-ophthalmologic abnormalities. A secondary form of MNGIE, called MNGIE without leukoencephalopathy, can be caused by mutations in the POLG gene.

3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.

Keutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein, MGP. Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP alleles will likely inherit KS.

Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.
Nakajo syndrome, also called nodular erythema with digital changes, is a rare autosomal recessive congenital disorder first reported in 1939 by A. Nakajo in the offspring of consanguineous parents. The syndrome can be characterized by erythema, loss of body fat in the upper part of the body, and disproportionately large eyes, ears, nose, lips, and fingers.
Stimmler syndrome is a rare autosomal recessive congenital disorder first described by Stimmler et al. in 1970. It is characterized by dwarfism, diabetes, a small head, and high levels of alanine in the urine.
Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.
Childhood cataract is cataract that occurs at birth or in childhood. It may be congenital or acquired.
Glomerulocystic kidney disease (GCKD) is a cystic disorder of the kidneys. GCKD involves cystic dilation of Bowman's capsule. It can occur with or without congenital abnormality.

Morse–Rawnsley–Sargent syndrome is an extraordinarily rare and deadly congenital malformation syndrome which affects the central nervous system during embryogenesis. It occurs before conception.

Hall-Riggs syndrome is a rare genetic disorder that causes neurological issues and birth defects. People with Hall-Riggs syndrome usually have skeletal dysplasia, facial deformities, and intellectual disabilities. Only 8 cases from 2 families worldwide have been described in medical literature. It is an autosomal recessive genetic disorder, meaning both parents must carry the gene in order for their offspring to be affected.