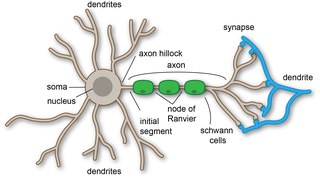
Dendrites, also dendrons, are branched protoplasmic extensions of a nerve cell that propagate the electrochemical stimulation received from other neural cells to the cell body, or soma, of the neuron from which the dendrites project. Electrical stimulation is transmitted onto dendrites by upstream neurons via synapses which are located at various points throughout the dendritic tree.

Chemical synapses are biological junctions through which neurons' signals can be sent to each other and to non-neuronal cells such as those in muscles or glands. Chemical synapses allow neurons to form circuits within the central nervous system. They are crucial to the biological computations that underlie perception and thought. They allow the nervous system to connect to and control other systems of the body.

A dendritic spine is a small membranous protrusion from a neuron's dendrite that typically receives input from a single axon at the synapse. Dendritic spines serve as a storage site for synaptic strength and help transmit electrical signals to the neuron's cell body. Most spines have a bulbous head, and a thin neck that connects the head of the spine to the shaft of the dendrite. The dendrites of a single neuron can contain hundreds to thousands of spines. In addition to spines providing an anatomical substrate for memory storage and synaptic transmission, they may also serve to increase the number of possible contacts between neurons. It has also been suggested that changes in the activity of neurons have a positive effect on spine morphology.

In neuroscience, long-term potentiation (LTP) is a persistent strengthening of synapses based on recent patterns of activity. These are patterns of synaptic activity that produce a long-lasting increase in signal transmission between two neurons. The opposite of LTP is long-term depression, which produces a long-lasting decrease in synaptic strength.
In neuroscience, synaptic plasticity is the ability of synapses to strengthen or weaken over time, in response to increases or decreases in their activity. Since memories are postulated to be represented by vastly interconnected neural circuits in the brain, synaptic plasticity is one of the important neurochemical foundations of learning and memory.
In neurophysiology, long-term depression (LTD) is an activity-dependent reduction in the efficacy of neuronal synapses lasting hours or longer following a long patterned stimulus. LTD occurs in many areas of the CNS with varying mechanisms depending upon brain region and developmental progress.

An excitatory synapse is a synapse in which an action potential in a presynaptic neuron increases the probability of an action potential occurring in a postsynaptic cell. Neurons form networks through which nerve impulses travel, each neuron often making numerous connections with other cells. These electrical signals may be excitatory or inhibitory, and, if the total of excitatory influences exceeds that of the inhibitory influences, the neuron will generate a new action potential at its axon hillock, thus transmitting the information to yet another cell.
Synaptogenesis is the formation of synapses between neurons in the nervous system. Although it occurs throughout a healthy person's lifespan, an explosion of synapse formation occurs during early brain development, known as exuberant synaptogenesis. Synaptogenesis is particularly important during an individual's critical period, during which there is a certain degree of synaptic pruning due to competition for neural growth factors by neurons and synapses. Processes that are not used, or inhibited during their critical period will fail to develop normally later on in life.
Schaffer collaterals are axon collaterals given off by CA3 pyramidal cells in the hippocampus. These collaterals project to area CA1 of the hippocampus and are an integral part of memory formation and the emotional network of the Papez circuit, and of the hippocampal trisynaptic loop. It is one of the most studied synapses in the world and named after the Hungarian anatomist-neurologist Károly Schaffer.
Metaplasticity is a term originally coined by W.C. Abraham and M.F. Bear to refer to the plasticity of synaptic plasticity. Until that time synaptic plasticity had referred to the plastic nature of individual synapses. However this new form referred to the plasticity of the plasticity itself, thus the term meta-plasticity. The idea is that the synapse's previous history of activity determines its current plasticity. This may play a role in some of the underlying mechanisms thought to be important in memory and learning such as long-term potentiation (LTP), long-term depression (LTD) and so forth. These mechanisms depend on current synaptic "state", as set by ongoing extrinsic influences such as the level of synaptic inhibition, the activity of modulatory afferents such as catecholamines, and the pool of hormones affecting the synapses under study. Recently, it has become clear that the prior history of synaptic activity is an additional variable that influences the synaptic state, and thereby the degree, of LTP or LTD produced by a given experimental protocol. In a sense, then, synaptic plasticity is governed by an activity-dependent plasticity of the synaptic state; such plasticity of synaptic plasticity has been termed metaplasticity. There is little known about metaplasticity, and there is much research currently underway on the subject, despite its difficulty of study, because of its theoretical importance in brain and cognitive science. Most research of this type is done via cultured hippocampus cells or hippocampal slices.

In the nervous system, a synapse is a structure that permits a neuron to pass an electrical or chemical signal to another neuron or to the target effector cell.
The synaptotropic hypothesis, also called the synaptotrophic hypothesis, is a neurobiological hypothesis of neuronal growth and synapse formation. The hypothesis was first formulated by J.E. Vaughn in 1988, and remains a focus of current research efforts. The synaptotropic hypothesis proposes that input from a presynaptic to a postsynaptic cell eventually can change the course of synapse formation at dendritic and axonal arbors. This synapse formation is required for the development of neuronal structure in the functioning brain.
Neural backpropagation is the phenomenon in which, after the action potential of a neuron creates a voltage spike down the axon, another impulse is generated from the soma and propagates towards the apical portions of the dendritic arbor or dendrites. In addition to active backpropagation of the action potential, there is also passive electrotonic spread. While there is ample evidence to prove the existence of backpropagating action potentials, the function of such action potentials and the extent to which they invade the most distal dendrites remain highly controversial.
Coincidence detection in the context of neurobiology is a process by which a neuron or a neural circuit can encode information by detecting the occurrence of temporally close but spatially distributed input signals. Coincidence detectors influence neuronal information processing by reducing temporal jitter, reducing spontaneous activity, and forming associations between separate neural events. This concept has led to a greater understanding of neural processes and the formation of computational maps in the brain.

Axon terminals are distal terminations of the telodendria (branches) of an axon. An axon, also called a nerve fiber, is a long, slender projection of a nerve cell, or neuron, that conducts electrical impulses called action potentials away from the neuron's cell body, or soma, in order to transmit those impulses to other neurons, muscle cells or glands.

Nonsynaptic plasticity is a form of neuroplasticity that involves modification of ion channel function in the axon, dendrites, and cell body that results in specific changes in the integration of excitatory postsynaptic potentials and inhibitory postsynaptic potentials. Nonsynaptic plasticity is a modification of the intrinsic excitability of the neuron. It interacts with synaptic plasticity, but it is considered a separate entity from synaptic plasticity. Intrinsic modification of the electrical properties of neurons plays a role in many aspects of plasticity from homeostatic plasticity to learning and memory itself. Nonsynaptic plasticity affects synaptic integration, subthreshold propagation, spike generation, and other fundamental mechanisms of neurons at the cellular level. These individual neuronal alterations can result in changes in higher brain function, especially learning and memory. However, as an emerging field in neuroscience, much of the knowledge about nonsynaptic plasticity is uncertain and still requires further investigation to better define its role in brain function and behavior.
Synaptic tagging, or the synaptic tagging hypothesis, was first proposed in 1997 by Uwe Frey and Richard G. Morris; it seeks to explain how neural signaling at a particular synapse creates a target for subsequent plasticity-related product (PRP) trafficking essential for sustained LTP and LTD. Although the molecular identity of the tags remains unknown, it has been established that they form as a result of high or low frequency stimulation, interact with incoming PRPs, and have a limited lifespan.
Long-term potentiation (LTP), thought to be the cellular basis for learning and memory, involves a specific signal transmission process that underlies synaptic plasticity. Among the many mechanisms responsible for the maintenance of synaptic plasticity is the cadherin–catenin complex. By forming complexes with intracellular catenin proteins, neural cadherins (N-cadherins) serve as a link between synaptic activity and synaptic plasticity, and play important roles in the processes of learning and memory.

Homosynaptic plasticity is one type of synaptic plasticity. Homosynaptic plasticity is input-specific, meaning changes in synapse strength occur only at post-synaptic targets specifically stimulated by a pre-synaptic target. Therefore, the spread of the signal from the pre-synaptic cell is localized.

Synaptic stabilization is crucial in the developing and adult nervous systems and is considered a result of the late phase of long-term potentiation (LTP). The mechanism involves strengthening and maintaining active synapses through increased expression of cytoskeletal and extracellular matrix elements and postsynaptic scaffold proteins, while pruning less active ones. For example, cell adhesion molecules (CAMs) play a large role in synaptic maintenance and stabilization. Gerald Edelman discovered CAMs and studied their function during development, which showed CAMs are required for cell migration and the formation of the entire nervous system. In the adult nervous system, CAMs play an integral role in synaptic plasticity relating to learning and memory.
