
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The process of analyzing and interpreting data can sometimes be referred to as computational biology, however this distinction between the two terms is often disputed. To some, the term computational biology refers to building and using models of biological systems.

Genomics is an interdisciplinary field of molecular biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.

Computational biology refers to the use of techniques in computer science, data analysis, mathematical modeling and computational simulations to understand biological systems and relationships. An intersection of computer science, biology, and data science, the field also has foundations in applied mathematics, molecular biology, cell biology, chemistry, and genetics.

Comparative genomics is a branch of biological research that examines genome sequences across a spectrum of species, spanning from humans and mice to a diverse array of organisms from bacteria to chimpanzees. This large-scale holistic approach compares two or more genomes to discover the similarities and differences between the genomes and to study the biology of the individual genomes. Comparison of whole genome sequences provides a highly detailed view of how organisms are related to each other at the gene level. By comparing whole genome sequences, researchers gain insights into genetic relationships between organisms and study evolutionary changes. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, Comparative genomics provides a powerful tool for studying evolutionary changes among organisms, helping to identify genes that are conserved or common among species, as well as genes that give unique characteristics of each organism. Moreover, these studies can be performed at different levels of the genomes to obtain multiple perspectives about the organisms.
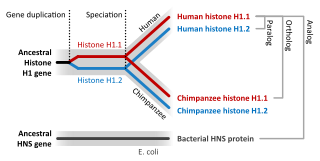
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

Michael Ashburner was an English biologist and Professor in the Department of Genetics at University of Cambridge. He also served as joint-head and co-founder of the European Bioinformatics Institute (EBI) of the European Molecular Biology Laboratory (EMBL) and a Fellow of Churchill College, Cambridge.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

John Frederick William Birney is joint director of EMBL's European Bioinformatics Institute (EMBL-EBI), in Hinxton, Cambridgeshire and deputy director general of the European Molecular Biology Laboratory (EMBL). He also serves as non-executive director of Genomics England, chair of the Global Alliance for Genomics and Health (GA4GH) and honorary professor of bioinformatics at the University of Cambridge. Birney has made significant contributions to genomics, through his development of innovative bioinformatics and computational biology tools. He previously served as an associate faculty member at the Wellcome Trust Sanger Institute.

Human accelerated regions (HARs), first described in August 2006, are a set of 49 segments of the human genome that are conserved throughout vertebrate evolution but are strikingly different in humans. They are named according to their degree of difference between humans and chimpanzees. Found by scanning through genomic databases of multiple species, some of these highly mutated areas may contribute to human-specific traits. Others may represent loss of functional mutations, possibly due to the action of biased gene conversion rather than adaptive evolution.

David Haussler is an American bioinformatician known for his work leading the team that assembled the first human genome sequence in the race to complete the Human Genome Project and subsequently for comparative genome analysis that deepens understanding the molecular function and evolution of the genome.
Anders Krogh is a bioinformatician at the University of Copenhagen, where he leads the university's bioinformatics center. He is known for his pioneering work on the use of hidden Markov models in bioinformatics, and is co-author of a widely used textbook in bioinformatics. In addition, he also co-authored one of the early textbooks on neural networks. His current research interests include promoter analysis, non-coding RNA, gene prediction and protein structure prediction.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
hCONDELs refer to regions of deletions within the human genome containing sequences that are highly conserved among closely related relatives. Almost all of these deletions fall within regions that perform non-coding functions. These represent a new class of regulatory sequences and may have played an important role in the development of specific traits and behavior that distinguish closely related organisms from each other.

Alfonso Valencia is a Spanish biologist, ICREA Professor, current director of the Life Sciences department at Barcelona Supercomputing Center, of Spanish National Bioinformatics Institute (INB-ISCIII), and coordinator of the data pillar of the Spanish Personalised Medicine initiative, IMPaCT. From 2015 to 2018, he was President of the International Society for Computational Biology.
Mathieu Daniel Blanchette is a computational biologist and Director of the School of Computer Science at McGill University. His research focuses on developing new algorithms for the detection of functional regions in DNA sequences.

Janet Kelso is a South African computational biologist and Group leader of the Minerva Research Group for Bioinformatics at the Max Planck Institute for Evolutionary Anthropology. She is best known for her work comparing DNA from previous humans with those of the present.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.
Katherine Snowden Pollard is the Director of the Gladstone Institute of Data Science and Biotechnology and a professor at the University of California, San Francisco (UCSF). She is a Chan Zuckerberg Biohub Investigator. She was awarded Fellowship of the International Society for Computational Biology in 2020 and the American Institute for Medical and Biological Engineering in 2021 for outstanding contributions to computational biology and bioinformatics.
