Hereditary haemochromatosis type 1 is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores. Humans, like most animals, have no mechanism to regulate excess iron, simply losing a limited amount through various means like sweating or menstruating.
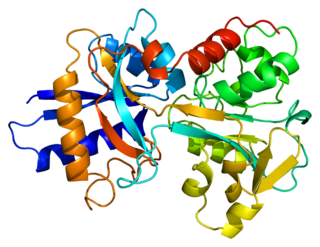
Transferrins are glycoproteins found in vertebrates which bind and consequently mediate the transport of iron (Fe) through blood plasma. They are produced in the liver and contain binding sites for two Fe3+ ions. Human transferrin is encoded by the TF gene and produced as a 76 kDa glycoprotein.

Iron overload is the abnormal and increased accumulation of total iron in the body, leading to organ damage. The primary mechanism of organ damage is oxidative stress, as elevated intracellular iron levels increase free radical formation via the Fenton reaction. Iron overload is often primary but may also be secondary to repeated blood transfusions. Iron deposition most commonly occurs in the liver, pancreas, skin, heart, and joints. People with iron overload classically present with the triad of liver cirrhosis, secondary diabetes mellitus, and bronze skin. However, due to earlier detection nowadays, symptoms are often limited to general chronic malaise, arthralgia, and hepatomegaly.
Microcytic anaemia is any of several types of anemia characterized by smaller than normal red blood cells. The normal mean corpuscular volume is approximately 80–100 fL. When the MCV is <80 fL, the red cells are described as microcytic and when >100 fL, macrocytic. The MCV is the average red blood cell size.
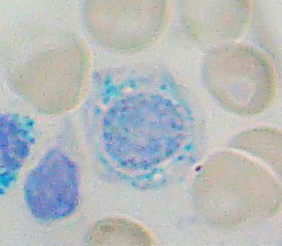
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.
Diamond–Blackfan anemia (DBA) is a congenital erythroid aplasia that usually presents in infancy. DBA causes low red blood cell counts (anemia), without substantially affecting the other blood components, which are usually normal. This is in contrast to Shwachman–Bodian–Diamond syndrome, in which the bone marrow defect results primarily in neutropenia, and Fanconi anemia, where all cell lines are affected resulting in pancytopenia. There is a risk to develop acute myelogenous leukemia (AML) and certain other cancers.

Total iron-binding capacity (TIBC) or sometimes transferrin iron-binding capacity is a medical laboratory test that measures the blood's capacity to bind iron with transferrin. Transferrin can bind two atoms of ferric iron (Fe3+) with high affinity. It means that transferrin has the capacity to transport approximately from 1.40 to 1.49 mg of iron per gram of transferrin present in the blood.
Transferrin saturation (TS), measured as a percentage, is a medical laboratory value. It is the value of serum iron divided by the total iron-binding capacity of the available transferrin, the main protein that binds iron in the blood, this value tells a clinician how much serum iron is bound. For instance, a value of 15% means that 15% of iron-binding sites of transferrin are being occupied by iron. The three results are usually reported together. A low transferrin saturation is a common indicator of iron deficiency anemia whereas a high transferrin saturation may indicate iron overload or hemochromatosis. Transferrin saturation is also called transferrin saturation index (TSI) or transferrin saturation percentage (TS%)

Hepcidin is a protein that in humans is encoded by the HAMP gene. Hepcidin is a key regulator of the entry of iron into the circulation in mammals.

Aceruloplasminemia is a rare autosomal recessive disorder in which the liver can not synthesize the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time.

Human homeostatic iron regulator protein, also known as the HFE protein, is a transmembrane protein that in humans is encoded by the HFE gene. The HFE gene is located on short arm of chromosome 6 at location 6p22.2

Hypochromic anemia is a generic term for any type of anemia in which the red blood cells are paler than normal. A normal red blood cell has a biconcave disk shape and will have an area of pallor in its center when viewed microscopically. In hypochromic cells, this area of central pallor is increased. This decrease in redness is due to a disproportionate reduction of red cell hemoglobin in proportion to the volume of the cell. Clinically the color can be evaluated by the mean corpuscular hemoglobin (MCH) or mean corpuscular hemoglobin concentration (MCHC). The MCHC is considered the better parameter of the two as it adjusts for effect the size of the cell has on its amount of hemoglobin. Hypochromia is clinically defined as below the normal MCH reference range of 27–33 picograms/cell in adults or below the normal MCHC reference range of 33–36 g/dL in adults.
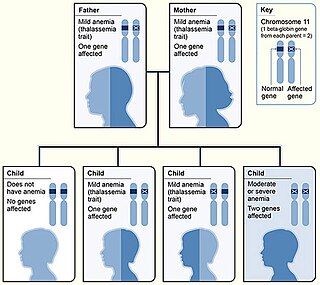
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.
Juvenile hemochromatosis, also known as hemochromatosis type 2, is a rare form of hereditary hemochromatosis, which emerges in young individuals, typically between 15 and 30 years of age, but occasionally later. It is characterized by an inability to control how much iron is absorbed by the body, in turn leading to iron overload, where excess iron accumulates in many areas of the body and causes damage to the places it accumulates.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.
Haemochromatosis type 3 is a type of iron overload disorder associated with deficiencies in transferrin receptor 2. It exhibits an autosomal recessive inheritance pattern. The first confirmed case was diagnosed in 1865 by French doctor Trousseau. Later in 1889, the German doctor von Recklinghausen indicated that the liver contains iron, and due to bleeding being considered to be the cause, he called the pigment "Haemochromatosis." In 1935, English doctor Sheldon's groundbreaking book titled, Haemochromatosis, reviewed 311 patient case reports and presented the idea that haemochromatosis was a congenital metabolic disorder. Hereditary haemochromatosis is a congenital disorder which affects the regulation of iron metabolism thus causing increased gut absorption of iron and a gradual build-up of pathologic iron deposits in the liver and other internal organs, joint capsules and the skin. The iron overload could potentially cause serious disease from the age of 40–50 years. In the final stages of the disease, the major symptoms include liver cirrhosis, diabetes and bronze-colored skin. There are four types of hereditary hemochromatosis which are classified depending on the age of onset and other factors such as genetic cause and mode of inheritance.

Congenital dyserythropoietic anemia (CDA) is a rare blood disorder, similar to the thalassemias. CDA is one of many types of anemia, characterized by ineffective erythropoiesis, and resulting from a decrease in the number of red blood cells (RBCs) in the body and a less than normal quantity of hemoglobin in the blood. CDA may be transmitted by both parents autosomal recessively or dominantly.
Congenital dyserythropoietic anemia type II, or hereditary erythroblastic multinuclearity with positive acidified serum lysis test (HEMPAS) is a rare genetic anemia in humans characterized by hereditary erythroblastic multinuclearity with positive acidified serum lysis test.
Hemochromatosis type 4 is a hereditary iron overload disorder that affects ferroportin, an iron transport protein needed to export iron from cells into circulation. Although the disease is rare, it is found throughout the world and affects people from various ethnic groups. While the majority of individuals with type 4 hemochromatosis have a relatively mild form of the disease, some affected individuals have a more severe form. As the disease progresses, iron may accumulate in the tissues of affected individuals over time, potentially resulting in organ damage.