In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.

A chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.

The Prato reaction is a particular example of the well-known 1,3-dipolar cycloaddition of azomethine ylides to olefins. In fullerene chemistry this reaction refers to the functionalization of fullerenes and nanotubes. The amino acid sarcosine reacts with paraformaldehyde when heated at reflux in toluene to an ylide which reacts with a double bond in a 6,6 ring position in a fullerene via a 1,3-dipolar cycloaddition to yield a N-methylpyrrolidine derivative or pyrrolidinofullerene or pyrrolidino[[3,4:1,2]] [60]fullerene in 82% yield based on C60 conversion.
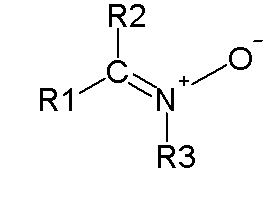
In organic chemistry, a 1,3-dipolar compound or 1,3-dipole is a dipolar compound with delocalized electrons and a separation of charge over three atoms. They are reactants in 1,3-dipolar cycloadditions.
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.

Aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine (-NR-) and two methylene bridges. The parent compound is aziridine, with molecular formula C
2H
4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
Isoindoline is a heterocyclic organic compound with the molecular formula C8H9N. The parent compound has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered nitrogen-containing ring. The compound's structure is similar to indoline except that the nitrogen atom is in the 2 position instead of the 1 position of the five-membered ring. Isoindoline itself is not commonly encountered, but several derivatives are found in nature and some synthetic derivatives are commercially valuable drugs, e.g. pazinaclone.
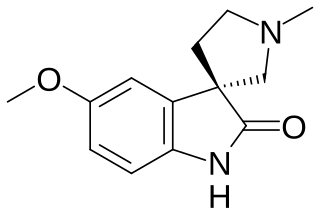
Horsfiline is an oxindole alkaloid found in the plant Horsfieldia superba, which is used in traditional herbal medicine. It has analgesic effects and has been the subject of research both to produce it synthetically by convenient routes and to develop analogues and derivatives which may have improved analgesic effects.
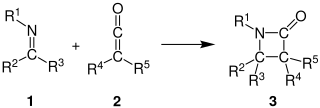
The Staudinger synthesis, also called the Staudinger ketene-imine cycloaddition, is a chemical synthesis in which an imine 1 reacts with a ketene 2 through a non-photochemical 2+2 cycloaddition to produce a β-lactam3. The reaction carries particular importance in the synthesis of β-Lactam antibiotics. The Staudinger synthesis should not be confused with the Staudinger reaction, a phosphine or phosphite reaction used to reduce azides to amines.
Nitrile ylides also known as nitrilium ylides, or nitrilium methylides are generally reactive intermediates. With a few exceptions, they cannot be isolated. However, a structure has been determined on a particularly stable nitrile ylide by X-ray crystallography. Another nitrile ylide has been captured under cryogenic conditions.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a [3+2] cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
The term bioorthogonal chemistry refers to any chemical reaction that can occur inside of living systems without interfering with native biochemical processes. The term was coined by Carolyn R. Bertozzi in 2003. Since its introduction, the concept of the bioorthogonal reaction has enabled the study of biomolecules such as glycans, proteins, and lipids in real time in living systems without cellular toxicity. A number of chemical ligation strategies have been developed that fulfill the requirements of bioorthogonality, including the 1,3-dipolar cycloaddition between azides and cyclooctynes, between nitrones and cyclooctynes, oxime/hydrazone formation from aldehydes and ketones, the tetrazine ligation, the isocyanide-based click reaction, and most recently, the quadricyclane ligation.
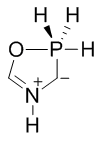
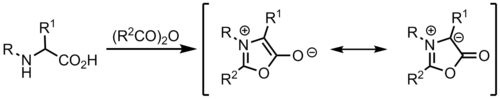
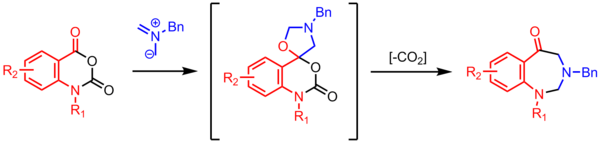
Montréalone is a mesoionic heterocyclic chemical compound. It is named for the city of Montréal, Canada, which is the location of McGill University, where it was first discovered.

Alkene carboamination is the simultaneous formation of C–N and C–C bonds across an alkene. This method represents a powerful strategy to build molecular complexity with up to two stereocenters in a single operation. Generally, there are four categories of reaction modes for alkene carboamination. The first class is cyclization reactions, which will form a N-heterocycle as a result. The second class has been well established in the last decade. Alkene substrates with a tethered nitrogen nucleophile have been used in these transformations to promote intramolecular aminocyclization. While intermolecular carboamination is extremely hard, people have developed a strategy to combine the nitrogen and carbon part, which is known as the third class. The most general carboamination, which takes three individual parts and couples them together is still underdeveloped.

Ferulic acid decarboxylases (Fdc) are decarboxylase enzymes capable of the reversible decarboxylation of aromatic carboxylic acids such as ferulic acid and cinnamic acid. Fdc's are fungal homologues of the E.coli UbiD enzyme which is involved in ubiquinone biosynthesis. This places Fdc within the wider UbiD enzyme family, representing a distinct clade within the family Presence of fdc1 and the associated pad1 genes were shown to be required for the decarboxylation of phenylacrylic acids in Saccharomyces cerevisiae.
An organic azide is organic compounds containing the azide (N3) functional group. Because of the hazards associated with their use, few azides are used commercially although they exhibit interesting reactivity for researchers. Low molecular weight azides are considered especially hazardous and are avoided. In the research laboratory, azides are precursors to amines. They are also popular for their participation in the "click reaction" and in Staudinger ligation. These two reactions are generally quite reliable, lending themselves to combinatorial chemistry.