
Amniocentesis is a medical procedure used primarily in the prenatal diagnosis of genetic conditions. It has other uses such as in the assessment of infection and fetal lung maturity. Prenatal diagnostic testing, which includes amniocentesis, is necessary to conclusively diagnose the majority of genetic disorders, with amniocentesis being the gold-standard procedure after 15 weeks' gestation.

Obstetric ultrasonography, or prenatal ultrasound, is the use of medical ultrasonography in pregnancy, in which sound waves are used to create real-time visual images of the developing embryo or fetus in the uterus (womb). The procedure is a standard part of prenatal care in many countries, as it can provide a variety of information about the health of the mother, the timing and progress of the pregnancy, and the health and development of the embryo or fetus.

A pulmonary sequestration is a medical condition wherein a piece of tissue that ultimately develops into lung tissue is not attached to the pulmonary arterial blood supply, as is the case in normally developing lung. This sequestered tissue is therefore not connected to the normal bronchial airway architecture, and fails to function in, and contribute to, respiration of the organism.

Hydrops fetalis or hydrops foetalis is a condition in the fetus characterized by an accumulation of fluid, or edema, in at least two fetal compartments. By comparison, hydrops allantois or hydrops amnion is an accumulation of excessive fluid in the allantoic or amniotic space, respectively.

Pulmonary atresia is a congenital malformation of the pulmonary valve in which the valve orifice fails to develop. The valve is completely closed thereby obstructing the outflow of blood from the heart to the lungs. The pulmonary valve is located on the right side of the heart between the right ventricle and pulmonary artery. In a normal functioning heart, the opening to the pulmonary valve has three flaps that open and close.

Lymphatic malformations are benign slow-flow type of vascular malformation of the lymphatic system characterized by lymphatic vessels which do not connect to the normal lymphatic circulation. The term lymphangioma is outdated and newer research reference the term lymphatic malformation.

Fetal surgery, also known as antenatal surgery or prenatal surgery, is a growing branch of maternal-fetal medicine that covers any of a broad range of surgical techniques that are used to treat congenital abnormalities in fetuses who are still in the pregnant uterus. There are three main types: open fetal surgery, which involves completely opening the uterus to operate on the fetus; minimally invasive fetoscopic surgery, which uses small incisions and is guided by fetoscopy and sonography; and percutaneous fetal therapy, which involves placing a catheter under continuous ultrasound guidance.

The EXIT procedure, or ex utero intrapartum treatment procedure, is a specialized surgical delivery procedure used to deliver babies who have airway compression. Causes of airway compression in newborn babies result from a number of rare congenital disorders, including bronchopulmonary sequestration, congenital cystic adenomatoid malformation, mouth or neck tumor such as teratoma, and lung or pleural tumor such as pleuropulmonary blastoma. Airway compression discovered at birth is a medical emergency. In many cases, however, the airway compression is discovered during prenatal ultrasound exams, permitting time to plan a safe delivery using the EXIT procedure or other means.
Sacrococcygeal teratoma (SCT) is a type of tumor known as a teratoma that develops at the base of the coccyx (tailbone) and is thought to be primarily derived from remnants of the primitive streak. Sacrococcygeal teratomas are benign 75% of the time, malignant 12% of the time, and the remainder are considered "immature teratomas" that share benign and malignant features. Benign sacrococcygeal teratomas are more likely to develop in younger children who are less than 5 months old, and older children are more likely to develop malignant sacrococcygeal teratomas.

Simpson–Golabi–Behmel syndrome (SGBS) is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.
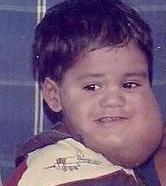
A cystic hygroma is a form of lymphatic malformation. It is an abnormal growth that usually appears on a baby's neck or head. It consists of one or more cysts and tends to grow larger over time. The disorder usually develops while the fetus is still in the uterus, but can also appear after birth.
Hemoglobin Barts, abbreviated Hb Barts, is an abnormal type of hemoglobin that consists of four gamma globins. It is moderately insoluble, and therefore accumulates in the red blood cells. Hb Barts has an extremely high affinity for oxygen, so it cannot release oxygen to the tissue. Therefore, this makes it an inefficient oxygen carrier. As an embryo develops, it begins to produce alpha-globins at weeks 5–6 of development. When both of the HBA1 and HBA2 genes which code for alpha globins becomes dysfunctional, the affected fetuses will have difficulty in synthesizing a functional hemoglobin. As a result, gamma chains will accumulate and form four gamma globins. These gamma globins bind to form hemoglobin Barts. It is produced in the disease alpha-thalassemia and in the most severe of cases, it is the only form of hemoglobin in circulation. In this situation, a fetus will develop hydrops fetalis and normally die before or shortly after birth, unless intrauterine blood transfusion is performed.
The Fetal Treatment Center at the University of California, San Francisco is a multidisciplinary care center dedicated to the diagnosis, treatment, and long-term follow-up of fetal birth defects. It combines the talents of specialists in pediatric surgery, genetics, obstetrics/perinatology, radiology, nursing, and neonatal medicine.

Pulmonary hypoplasia is an incomplete development of the lungs, resulting in an abnormally low number or small size of bronchopulmonary segments or alveoli. A congenital malformation, most often occurs secondary to other fetal abnormalities that interfere with normal development of the lungs. Primary (idiopathic) pulmonary hypoplasia is rare and usually not associated with other maternal or fetal abnormalities.
Congenital varicella syndrome is a rare disease resulting from Varicella Zoster virus (VZV) infection during the period of gestation. Viremia during the primary infection can result in transplacental transmission of the infection to the developing fetus. An estimated 25% of fetuses get infected with varicella infection when mother has a varicella infection during the pregnancy but the risk of developing congenital varicella syndrome is around 2%, therefore majority of the outcomes are normal newborns. Patients with primary infection before 20 weeks of gestation are at a higher risk of developing the severe form of infection, affecting the eyes, limbs, skin and the central nervous system. Diagnosis requires a documented history of primary infection in the mother and serial ultrasound demonstrating features suggestive of congenital varicella syndrome. There is no definitive treatment, termination of pregnancy in fetuses with severe features is recommended. Vaccination to prevent maternal varicella infection and proper counseling to avoid contact with infected people are important for the management options to reduce the incidence of congenital varicella syndrome.
Tracheal agenesis is a rare birth defect with a prevalence of less than 1 in 50,000 in which the trachea fails to develop, resulting in an impaired communication between the larynx and the alveoli of the lungs. Although the defect is normally fatal, occasional cases have been reported of long-term survival following surgical intervention.

Mediastinal shift is an abnormal movement of the mediastinal structures toward one side of the chest cavity. A shift indicates a severe imbalance of pressures inside the chest. Mediastinal shifts are generally caused by increased lung volume, decreased lung volume, or abnormalities in the pleural space. Additionally, masses inside the mediastinum or musculoskeletal abnormalities can also lead to abnormal mediastinal arrangement. Typically, these shifts are observed on x-ray but also on computed tomography (CT) or magnetic resonance imaging (MRI). On chest x-ray, tracheal deviation, or movement of the trachea away from its midline position can be used as a sign of a shift. Other structures, like the heart, can also be used as reference points. Below are examples of pathologies that can cause a mediastinal shift and their appearance.

A lung cavity or pulmonary cavity is an abnormal, thick-walled, air-filled space within the lung. Cavities in the lung can be caused by infections, cancer, autoimmune conditions, trauma, congenital defects, or pulmonary embolism. The most common cause of a single lung cavity is lung cancer. Bacterial, mycobacterial, and fungal infections are common causes of lung cavities. Globally, tuberculosis is likely the most common infectious cause of lung cavities. Less commonly, parasitic infections can cause cavities. Viral infections almost never cause cavities. The terms cavity and cyst are frequently used interchangeably; however, a cavity is thick walled, while a cyst is thin walled. The distinction is important because cystic lesions are unlikely to be cancer, while cavitary lesions are often caused by cancer.
Epignathus is a rare teratoma of the oropharynx. Epignathus is a form of oropharyngeal teratoma that arises from the palate and, in most cases, results in death. The pathology is thought to be due to unorganized and uncontrolled differentiation of somatic cells leading to formation of the teratoma; sometimes it is also referred to as fetus in fetu, which is an extremely rare occurrence of an incomplete but parasitic fetus located in the body of its twin. This tumor is considered benign (non-cancerous) but life-threatening because of its atypical features and high risk of airway obstruction, which is the cause of death in 80-100% of the cases at the time of delivery.

Pulmonary agenesis is an inborn lung underdevelopment that is rare and potentially lethal. The disorder is caused by a complete developmental arrest of the primitive lung during embryonic life, and it is often associated with other developmental defects. Bilateral and unilateral pulmonary agenesis are classified, depending on whether one side of the lung or both sides are affected. Bilateral pulmonary agenesis is lethal, while the mortality rate of unilateral pulmonary agenesis is higher than 50%. Depending on the severity, the symptom ranges from none to various respiratory complaints. It is detectable prenatally, however, its nonspecific clinical features act as the obstacle for diagnosing. The exact cause of pulmonary agenesis is still obscure. However, theories have been raised regarding the vascular, iatrogenic, viral and genetic causes of pulmonary agenesis in an attempt to explain the pathogenesis of the disorder. In most cases of pulmonary agenesis, surgical resection is performed to remove the malformed lobe or the entire defected lung of the patient depending on the severity of the respiratory impairment.



