Barth syndrome (BTHS) is a rare but serious X-linked genetic disorder, caused by changes in phospholipid structure and metabolism. It may affect multiple body systems, and is potentially fatal. The syndrome is diagnosed almost exclusively in males.
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.

Wolfram syndrome, also called DIDMOAD, is a rare autosomal-recessive genetic disorder that causes childhood-onset diabetes mellitus, optic atrophy, and deafness as well as various other possible disorders including neurodegeneration. Symptoms can start to appear as early as childhood to adult years. There is a 25% recurrence risk in children.

Salla disease (SD) or mild Free Sialic Acid Storage Disease (FSASD) is an autosomal recessive lysosomal storage disease characterized by early physical impairment and intellectual disability. Salla disease was first reported as a lysosomal storage disorder in a family from northern Finland. Salla refers to the area where the affected family resided. It was first described in 1979, after Salla, a municipality in Finnish Lapland and is one of 40 Finnish heritage diseases. The term Salla disease is now used in the literature not only for FSASD cases with the Finnish founder variant in SLC17A5, but also for any mild FSASD cases, independent of the mutation or region of origin.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.

2-hydroxyglutaric aciduria is a rare neurometabolic disorder characterized by the significantly elevated levels of hydroxyglutaric acid in one's urine. It is either autosomal recessive or autosomal dominant.
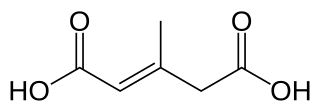
3-Methylglutaconic aciduria (MGA) is any of at least five metabolic disorders that impair the body's ability to make energy in the mitochondria. As a result of this impairment, 3-methylglutaconic acid and 3-methylglutaric acid build up and can be detected in the urine.
Dominant optic atrophy (DOA), or autosomal dominant optic atrophy (ADOA), (Kjer's type) is an autosomally inherited disease that affects the optic nerves, causing reduced visual acuity and blindness beginning in childhood. However, the disease can seem to re-present a second time with further vision loss due to the early onset of presbyopia symptoms (i.e., difficulty in viewing objects up close). DOA is characterized as affecting neurons called retinal ganglion cells (RGCs). This condition is due to mitochondrial dysfunction mediating the death of optic nerve fibers. The RGCs axons form the optic nerve. Therefore, the disease can be considered of the central nervous system. Dominant optic atrophy was first described clinically by Batten in 1896 and named Kjer’s optic neuropathy in 1959 after Danish ophthalmologist Poul Kjer, who studied 19 families with the disease. Although dominant optic atrophy is the most common autosomally inherited optic neuropathy (i.e., disease of the optic nerves), it is often misdiagnosed.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
MT-ATP6 is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 6' that encodes the ATP synthase Fo subunit 6. This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase. This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP. Mutations in the MT-ATP6 gene have been found in approximately 10 to 20 percent of people with Leigh syndrome.

Mitochondrial import inner membrane translocase subunit Tim8 A, also known as deafness-dystonia peptide or protein is an enzyme that in humans is encoded by the TIMM8A gene. This translocase has similarity to yeast mitochondrial proteins that are involved in the import of metabolite transporters from the cytoplasm into the mitochondrial inner membrane. The gene is mutated in deafness-dystonia syndrome and it is postulated that MTS/DFN-1 is a mitochondrial disease caused by a defective mitochondrial protein import system.

Optic atrophy 3 protein is a protein that in humans is encoded by the OPA3 gene.

Caseinolytic peptidase B protein homolog (CLPB), also known as Skd3, is a mitochondrial AAA ATPase chaperone that in humans is encoded by the gene CLPB, which encodes an adenosine triphosphate-(ATP) dependent chaperone. Skd3 is localized in mitochondria and widely expressed in human tissues. High expression in adult brain and low expression in granulocyte is found. It is a potent protein disaggregase that chaperones the mitochondrial intermembrane space. Mutations in the CLPB gene could cause autosomal recessive metabolic disorder with intellectual disability/developmental delay, congenital neutropenia, progressive brain atrophy, movement disorder, cataracts, and 3-methylglutaconic aciduria. Recently, heterozygous, dominant negative mutations in CLPB have been identified as a cause of severe congenital neutropenia (SCN).
Mitochondrial import inner membrane translocase subunit TIM50 is a protein that in humans is encoded by the TIMM50 gene. Tim50 is a subunit of the Tim23 translocase complex in the inner mitochondrial membrane. Mutations in TIMM50 can lead to epilepsy, severe intellectual disability, and 3-methylglutaconic aciduria. TIMM50 expression is increased in breast cancer cells and decreased in hypertrophic hearts.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.

Serine active site-containing protein 1, or Protein SERAC1 is a protein in humans that is encoded by the SERAC1 gene. The protein encoded by this gene is a phosphatidylglycerol remodeling protein found at the interface of mitochondria and endoplasmic reticula, where it mediates phospholipid exchange. The encoded protein plays a major role in mitochondrial function and intracellular cholesterol trafficking. Defects in this gene are a cause of 3-methylglutaconic aciduria with deafness, encephalopathy, and Leigh-like syndrome (MEGDEL). Two transcript variants, one protein-coding and the other non-protein coding, have been found for this gene.

Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.
Taosheng Huang is a physician-scientist with substantial academic achievements and professional experience in translational research, specifically, in human mitochondrial genetics. He is a full Professor and Director of the Molecular Diagnostic Laboratory in the Division of Human Genetics at Cincinnati Children’s Hospital Medical Center (CCHMC). Huang has published over 100 manuscripts in many impactful journals.
Proud syndrome is a very rare genetic disorder which is characterized by severe intellectual disabilities, corpus callosum agenesis, epilepsy, and spasticity. It is a type of syndromic X-linked intellectual disability.

Wolfram-like syndrome is a rare autosomal dominant genetic disorder that shares some of the features shown by those affected with the autosomal recessive Wolfram syndrome. It is a type of WFS1-related disorder.
