
In chemistry, hydrophobicity is the physical property of a molecule that is seemingly repelled from a mass of water. In contrast, hydrophiles are attracted to water.
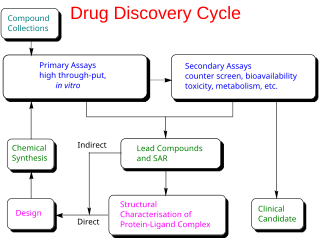
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
In the physical sciences, a partition coefficient (P) or distribution coefficient (D) is the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium. This ratio is therefore a comparison of the solubilities of the solute in these two liquids. The partition coefficient generally refers to the concentration ratio of un-ionized species of compound, whereas the distribution coefficient refers to the concentration ratio of all species of the compound.

Lipinski's rule of five, also known as Pfizer's rule of five or simply the rule of five (RO5), is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has chemical properties and physical properties that would likely make it a orally active drug in humans. The rule was formulated by Christopher A. Lipinski in 1997, based on the observation that most orally administered drugs are relatively small and moderately lipophilic molecules.
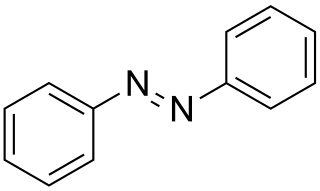
Azobenzene is a photoswitchable chemical compound composed of two phenyl rings linked by a N=N double bond. It is the simplest example of an aryl azo compound. The term 'azobenzene' or simply 'azo' is often used to refer to a wide class of similar compounds. These azo compounds are considered as derivatives of diazene (diimide), and are sometimes referred to as 'diazenes'. The diazenes absorb light strongly and are common dyes.

In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.

The Journal of Physical Chemistry A is a scientific journal which reports research on the chemistry of molecules - including their dynamics, spectroscopy, kinetics, structure, bonding, and quantum chemistry. It is published weekly by the American Chemical Society.

An amphiphile, or amphipath, is a chemical compound possessing both hydrophilic and lipophilic (fat-loving) properties. Such a compound is called amphiphilic or amphipathic. Common amphiphilic compound is surfactant, which is a main ingredient of soaps, detergents, and lipoproteins. The phospholipid amphiphiles are the major structural component of cell membranes.
Jaguar is a computer software package used for ab initio quantum chemistry calculations for both gas and solution phases. It is commercial software marketed by the company Schrödinger. The program was originated in research groups of Richard Friesner and William Goddard and was initially called PS-GVB.
In quantum and theoretical chemistry, an intruder state is a particular situation arising in perturbative evaluations, where the energy of the perturbers is comparable in magnitude to the energy associated to the zero order wavefunction. In this case, a divergent behavior occurs, due to the nearly zero denominator in the expression of the perturbative correction.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Druglikeness is a qualitative concept used in drug design for how "druglike" a substance is with respect to factors like bioavailability. It is estimated from the molecular structure before the substance is even synthesized and tested. A druglike molecule has properties such as:
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.

In chemistry, a water cluster is a discrete hydrogen bonded assembly or cluster of molecules of water. Many such clusters have been predicted by theoretical models (in silico), and some have been detected experimentally in various contexts such as ice, bulk liquid water, in the gas phase, in dilute mixtures with non-polar solvents, and as water of hydration in crystal lattices. The simplest example is the water dimer (H2O)2.
Electromerism is a type of isomerism between a pair of molecules differing in the way electrons are distributed among the atoms and the connecting chemical bonds. In some literature electromerism is equated to valence tautomerism, a term usually reserved for tautomerism involving reconnecting chemical bonds.
Minnesota Functionals (Myz) are a group of highly parameterized approximate exchange-correlation energy functionals in density functional theory (DFT). They are developed by the group of Prof. Donald Truhlar at the University of Minnesota. These functionals are based on the meta-GGA approximation, i.e. they include terms that depend on the kinetic energy density, and are all based on complicated functional forms parametrized on high-quality benchmark databases. These functionals can be used for traditional quantum chemistry and solid-state physics calculations. The Myz functionals are widely used and tested in the quantum chemistry community.
Dithionous acid is a sulfur oxoacid with the chemical formula H2S2O4. It is unstable in pure form, but its salts, known as dithionites, are stable.
Neon compounds are chemical compounds containing the element neon (Ne) with other molecules or elements from the periodic table. Compounds of the noble gas neon were believed not to exist, but there are now known to be molecular ions containing neon, as well as temporary excited neon-containing molecules called excimers. Several neutral neon molecules have also been predicted to be stable, but are yet to be discovered in nature. Neon has been shown to crystallize with other substances and form clathrates or Van der Waals solids.
A heterocumulene is a molecule or ion containing a chain of at least three double bonds between consecutive atoms, in which one or more atoms in the doubly bonded chain is a heteroatom. Such species are analogous to a cumulene in which the chain of doubly bonded atoms contains only carbon, except that at least one carbon is replaced by a heteroatom. Some authors relax the definition to include species with chains of only two double bonds between consecutive atoms, also known as heteroallenes.
Organoargon chemistry describes the synthesis and properties of chemical compounds containing a carbon to argon chemical bond.

