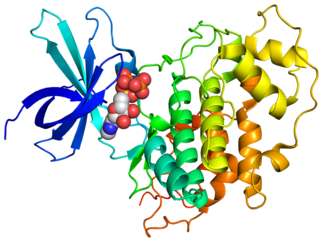
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine protein kinase that mediates the addition of phosphate molecules onto serine and threonine amino acid residues. First discovered in 1980 as a regulatory kinase for its namesake, glycogen synthase (GS), GSK-3 has since been identified as a protein kinase for over 100 different proteins in a variety of different pathways. In mammals, including humans, GSK-3 exists in two isozymes encoded by two homologous genes GSK-3α (GSK3A) and GSK-3β (GSK3B). GSK-3 has been the subject of much research since it has been implicated in a number of diseases, including type 2 diabetes, Alzheimer's disease, inflammation, cancer, and bipolar disorder.

Thromboxane A synthase 1 , also known as TBXAS1, is a cytochrome P450 enzyme that, in humans, is encoded by the TBXAS1 gene.

Enprostil is a synthetic prostaglandin designed to resemble dinoprostone. Enprostil was found to be a highly potent inhibitor of gastric HCl secretion. It is an analog of prostaglandin E2 but unlike this prostaglandin, which binds to and activates all four cellular receptors viz., EP1, EP2, EP3, and EP4 receptors, enprostil is a more selective receptor agonist in that it binds to and activates primarily the EP3 receptor. Consequently, enprostil is expected to have a narrower range of actions that may avoid some of the unwanted side-effects and toxicities of prostaglandin E2. A prospective multicenter randomized controlled trial conducted in Japan found combining enprostil with cimetidine was more effective than cimetidine alone in treating gastric ulcer.

Helenalin, or (-)-4-Hydroxy-4a,8-dimethyl-3,3a,4a,7a,8,9,9a-octahydroazuleno[6,5-b]furan-2,5-dione, is a toxic sesquiterpene lactone which can be found in several plants such as Arnica montana and Arnica chamissonis subsp. foliosa. Helenalin is responsible for the toxicity of the Arnica spp. Although toxic, helenalin possesses some in vitro anti-inflammatory and anti-neoplastic effects. Helenalin can inhibit certain enzymes, such as 5-lipoxygenase and leukotriene C4 synthase. For this reason the compound or its derivatives may have potential medical applications.

Benzofuranylpropylaminopentane is a drug with an unusual effects profile. It can loosely be grouped with the stimulant or antidepressant drug families, but its mechanism of action is quite different.

8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, PD-116,948) is a drug which acts as a potent and selective antagonist for the adenosine A1 receptor. It has high selectivity for A1 over other adenosine receptor subtypes, but as with other xanthine derivatives DPCPX also acts as a phosphodiesterase inhibitor, and is almost as potent as rolipram at inhibiting PDE4. It has been used to study the function of the adenosine A1 receptor in animals, which has been found to be involved in several important functions such as regulation of breathing and activity in various regions of the brain, and DPCPX has also been shown to produce behavioural effects such as increasing the hallucinogen-appropriate responding produced by the 5-HT2A agonist DOI, and the dopamine release induced by MDMA, as well as having interactions with a range of anticonvulsant drugs.

Diclofensine was developed by Hoffmann-La Roche in the 1970s in the search for a new antidepressant. It was found that the (S)-isomer was responsible for activity. Is a stimulant drug which acts as a triple monoamine reuptake inhibitor, primarily inhibiting the reuptake of dopamine and norepinephrine, with affinities (Ki) of 16.8 nM, 15.7 nM, and 51 nM for DAT, NET, and SERT, respectively. It was found to be an effective antidepressant in human trials, with relatively few side effects, but was ultimately dropped from clinical development, possibly due to concerns about its abuse potential.

4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) is an anion exchange inhibitor, blocking reversibly, and later irreversibly, exchangers such as chloride-bicarbonate exchanger.

Oxaprotiline, also known as hydroxymaprotiline, is a norepinephrine reuptake inhibitor of the tetracyclic antidepressant (TeCA) family that is related to maprotiline. Though investigated as an antidepressant, it was never marketed.

Nitroarginine, or Nω-nitro-l-arginine, also known as L-NOARG, is a nitro derivative of the amino acid arginine. It is an inhibitor of nitric oxide synthase and hence a vasoconstrictor. As such, it finds widespread use as a biochemical tool in the study of nitric oxide and its biological effects.

Tandamine is a selective norepinephrine reuptake inhibitor with a tricyclic structure. It was developed in the 1970s as an antidepressant but was never commercialized. Tandamine is analogous to pirandamine, which, instead, acts as a selective serotonin reuptake inhibitor (SSRI).

Isopimaric acid (IPA) is a toxin which acts as a large conductance Ca2+-activated K+ channel (BK channel) opener.

6-Nitroquipazine is a potent and selective serotonin reuptake inhibitor used in scientific research.
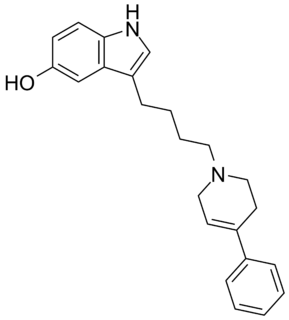
Roxindole (EMD-49,980) is a dopaminergic and serotonergic drug which was originally developed by Merck KGaA for the treatment of schizophrenia. In clinical trials its antipsychotic efficacy was only modest but it was unexpectedly found to produce potent and rapid antidepressant and anxiolytic effects. As a result, roxindole was further researched for the treatment of depression instead. It has also been investigated as a therapy for Parkinson's disease and prolactinoma.
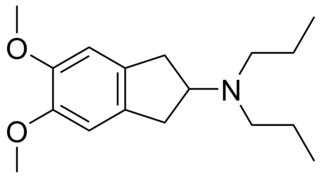
PNU-99,194(A) (or U-99,194(A)) is a drug which acts as a moderately selective D3 receptor antagonist with ~15-30-fold preference for D3 over the D2 subtype. Though it has substantially greater preference for D3 over D2, the latter receptor does still play some role in its effects, as evidenced by the fact that PNU-99,194 weakly stimulates both prolactin secretion and striatal dopamine synthesis, actions it does not share with the more selective (100-fold) D3 receptor antagonists S-14,297 and GR-103,691.

CGS-15943 is a drug which acts as a potent and reasonably selective antagonist for the adenosine receptors A1 and A2A, having a Ki of 3.3nM at A2A and 21nM at A1. It was one of the first adenosine receptor antagonists discovered that is not a xanthine derivative, instead being a triazoloquinazoline. Consequently, CGS-15943 has the advantage over most xanthine derivatives that it is not a phosphodiesterase inhibitor, and so has more a specific pharmacological effects profile. It produces similar effects to caffeine in animal studies, though with higher potency.

8-Cyclopentyl-1,3-dimethylxanthine (8-Cyclopentyltheophylline, 8-CPT, CPX) is a drug which acts as a potent and selective antagonist for the adenosine receptors, with some selectivity for the A1 receptor subtype, as well as a non-selective phosphodiesterase inhibitor. It has stimulant effects in animals with slightly higher potency than caffeine.
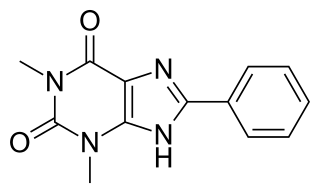
8-Phenyltheophylline (8-phenyl-1,3-dimethylxanthine, 8-PT) is a drug derived from the xanthine family which acts as a potent and selective antagonist for the adenosine receptors A1 and A2A, but unlike other xanthine derivatives has virtually no activity as a phosphodiesterase inhibitor. It has stimulant effects in animals with similar potency to caffeine. Coincidentally 8-phenyltheophylline has also been found to be a potent and selective inhibitor of the liver enzyme CYP1A2 which makes it likely to cause interactions with other drugs which are normally metabolised by CYP1A2.

LY-2183240 is a drug which acts both as a potent inhibitor of the reuptake of the endocannabinoid anandamide and as an inhibitor of fatty acid amide hydrolase (FAAH), the primary enzyme responsible for degrading anandamide. This leads to markedly elevated anandamide levels in the brain, and LY-2183240 has been shown to produce both analgesic and anxiolytic effects in animal models. While LY-2183240 is a potent inhibitor of FAAH, it has relatively poor selectivity and also inhibits several other enzyme side targets. Consequently, it was never developed for clinical use, though it remains widely used in research, and has also been sold as a designer drug.
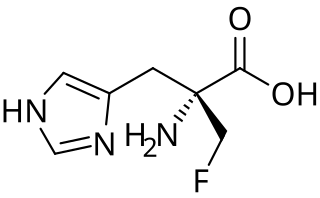
α-Fluoromethylhistidine (α-FMH) is an irreversible specific inhibitor of histidine decarboxylase (HDC). It functions by forming a covalent linkage with a catalytic serine residue on the active site of HDC. Due to its efficacy in reducing histamine levels in tissue mast cells, it has many applications in the study of histaminergic systems.
