
Tay–Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. The most common form is infantile Tay–Sachs disease, which becomes apparent around the age of three to six months of age, with the baby losing the ability to turn over, sit, or crawl. This is then followed by seizures, hearing loss, and inability to move, with death usually occurring by the age of three to five. Less commonly, the disease may occur later in childhood, adolescence, or adulthood. These forms tend to be less severe, but the juvenile form typically results in death by age 15.

The Portuguese Water Dog originated from the Algarve region of Portugal. From there the breed expanded to all around Portugal's coast, where they were taught to herd fish into fishermen's nets, retrieve lost tackle or broken nets, and act as couriers from ship to ship, or ship to shore. Portuguese Water Dogs rode in fishing trawlers as they worked their way from the Atlantic waters of Portugal to the waters off the coast of Iceland fishing for cod.

A ganglioside is a molecule composed of a glycosphingolipid with one or more sialic acids linked on the sugar chain. NeuNAc, an acetylated derivative of the carbohydrate sialic acid, makes the head groups of gangliosides anionic at pH 7, which distinguishes them from globosides.

Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B. These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues, and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death. The rare autosomal recessive neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.

GM2-gangliosidosis, AB variant is a rare, autosomal recessive metabolic disorder that causes progressive destruction of nerve cells in the brain and spinal cord. It has a similar pathology to Sandhoff disease and Tay–Sachs disease. The three diseases are classified together as the GM2 gangliosidoses, because each disease represents a distinct molecular point of failure in the activation of the same enzyme, beta-hexosaminidase. AB variant is caused by a failure in the gene that makes an enzyme cofactor for beta-hexosaminidase, called the GM2 activator.
A lipid storage disorder is any one of a group of inherited metabolic disorders in which harmful amounts of fats or lipids accumulate in some body cells and tissues. People with these disorders either do not produce enough of one of the enzymes needed to metabolize and break down lipids or, they produce enzymes that do not work properly. Over time, the buildup of fats may cause permanent cellular and tissue damage, particularly in the brain, peripheral nervous system, liver, spleen, and bone marrow.

Sphingolipidoses are a class of lipid storage disorders or degenerative storage disorders caused by deficiency of an enzyme that is required for the catabolism of lipids that contain ceramide, also relating to sphingolipid metabolism. The main members of this group are Niemann–Pick disease, Fabry disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and metachromatic leukodystrophy. They are generally inherited in an autosomal recessive fashion, but notably Fabry disease is X-linked recessive. Taken together, sphingolipidoses have an incidence of approximately 1 in 10,000, but substantially more in certain populations such as Ashkenazi Jews. Enzyme replacement therapy is available to treat mainly Fabry disease and Gaucher disease, and people with these types of sphingolipidoses may live well into adulthood. The other types are generally fatal by age 1 to 5 years for infantile forms, but progression may be mild for juvenile- or adult-onset forms.
The GM2 gangliosidoses are a group of three related genetic disorders that result from a deficiency of the enzyme beta-hexosaminidase. This enzyme catalyzes the biodegradation of fatty acid derivatives known as gangliosides. The diseases are better known by their individual names: Tay–Sachs disease, AB variant, and Sandhoff disease.
The GM1 gangliosidoses, usually shortened to GM1, are gangliosidoses caused by mutation in the GLB1 gene resulting in a deficiency of beta-galactosidase. The deficiency causes abnormal storage of acidic lipid materials in cells of the central and peripheral nervous systems, but particularly in the nerve cells, resulting in progressive neurodegeneration. GM1 is a rare lysosomal storage disorder with a prevalence of 1 to every 100,000 to 200,000 live births worldwide, although rates are higher in some regions.
GM1 (monosialotetrahexosylganglioside) the "prototype" ganglioside, is a member of the ganglio series of gangliosides which contain one sialic acid residue. GM1 has important physiological properties and impacts neuronal plasticity and repair mechanisms, and the release of neurotrophins in the brain. Besides its function in the physiology of the brain, GM1 acts as the site of binding for both cholera toxin and E. coli heat-labile enterotoxin.

Hexosaminidase is an enzyme involved in the hydrolysis of terminal N-acetyl-D-hexosamine residues in N-acetyl-β-D-hexosaminides.

Beta-hexosaminidase subunit beta is an enzyme that in humans is encoded by the HEXB gene.

GM2 ganglioside activator also known as GM2A is a protein which in humans is encoded by the GM2A gene.

Hexosaminidase A (alpha polypeptide), also known as HEXA, is an enzyme that in humans is encoded by the HEXA gene, located on the 15th chromosome.
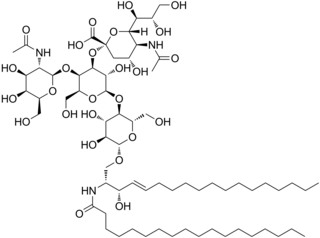
In organic chemistry, GM2 is a type of ganglioside. G refers to ganglioside, the M is for monosialic, and 2 refers to the fact that it was the second monosialic ganglioside discovered. It is associated with GM2 gangliosidoses such as Tay–Sachs disease.

A cherry-red spot is a finding in the macula of the eye in a variety of lipid storage disorders and in central retinal artery occlusion. It describes the appearance of a small circular choroid shape as seen through the fovea centralis. Its appearance is due to a relative transparency of the macula; storage disorders cause the accumulation of storage material within the cell layers of the retina, however, the macula, which is relatively devoid of cellular layers, does not build up this material, and thus allows the eye to see through the macula to the red choroid below.
Substrate presentation is a biological process that activates a protein. The protein is sequestered away from its substrate and then activated by release and exposure of the protein to its substrate. A substrate is typically the substance on which an enzyme acts but can also be a protein surface to which a ligand binds. The substrate is the material acted upon. In the case of an interaction with an enzyme, the protein or organic substrate typically changes chemical form. Substrate presentation differs from allosteric regulation in that the enzyme need not change its conformation to begin catalysis. Substrate presentation is best described for domain partitioning at nanoscopic distances (<100 nm).
PIP2 domains are a type of cholesterol-independent lipid domain formed from phosphatidylinositol and positively charged proteins in the plasma membrane. They tend to inhibit GM1 lipid raft function.

Levacetylleucine, sold under the brand name Aqneursa, is a medication used for the treatment of neurological manifestations of Niemann-Pick disease type C. Levacetylleucine is a modified version of the amino acid leucine (N-Acetyl-L-Leucine). It is taken by mouth.