
Hemoglobinopathy is the medical term for a group of inherited blood disorders involving the hemoglobin, the protein of red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time. Thalassemia is also known as Cooley's anemia or Mediterranean anemia.
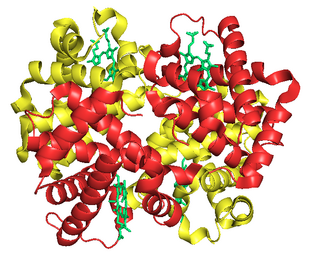
Fetal hemoglobin, or foetal haemoglobin is the main oxygen carrier protein in the human fetus. Hemoglobin F is found in fetal red blood cells, and is involved in transporting oxygen from the mother's bloodstream to organs and tissues in the fetus. It is produced at around 6 weeks of pregnancy and the levels remain high after birth until the baby is roughly 2–4 months old. Hemoglobin F has a different composition than adult forms of hemoglobin, allowing it to bind oxygen more strongly; this in turn enables the developing fetus to retrieve oxygen from the mother's bloodstream, which occurs through the placenta found in the mother's uterus.
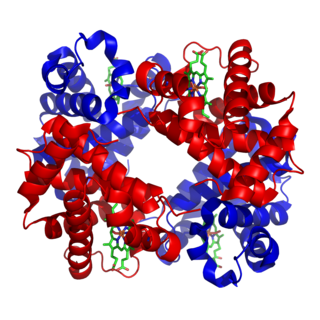
Hemoglobin A (HbA), also known as adult hemoglobin, hemoglobin A1 or α2β2, is the most common human hemoglobin tetramer, accounting for over 97% of the total red blood cell hemoglobin. Hemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs to the tissues. Hemoglobin A is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2). Hemoglobin A2 (HbA2) is a less common adult form of hemoglobin and is composed of two alpha and two delta-globin subunits. This hemoglobin makes up 1-3% of hemoglobin in adults.
Hemoglobin C is an abnormal hemoglobin in which glutamic acid residue at the 6th position of the β-globin chain is replaced with a lysine residue due to a point mutation in the HBB gene. People with one copy of the gene for hemoglobin C do not experience symptoms, but can pass the abnormal gene on to their children. Those with two copies of the gene are said to have hemoglobin C disease and can experience mild anemia. It is possible for a person to have both the gene for hemoglobin S and the gene for hemoglobin C; this state is called hemoglobin SC disease, and is generally more severe than hemoglobin C disease, but milder than sickle cell anemia.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.
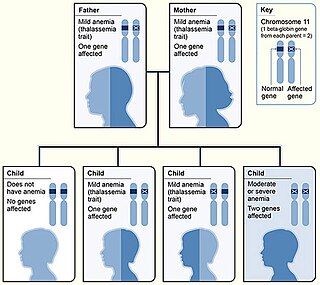
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Sickle cell disease (SCD), also simply called sickle cell, is a group of hemoglobin-related blood disorders typically inherited. The most common type is known as sickle cell anemia. It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells. This leads to a rigid, sickle-like shape under certain circumstances. Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain in joints, anemia, swelling in the hands and feet, bacterial infections, dizziness and stroke. Long-term pain may develop as people get older. The average life expectancy in the developed world is 40 to 60 years. It often gets worse within age. All the major organs are affected by sickle cell disease. The liver, heart, kidneys, gallbladder, eyes, bones, and joints also can suffer damage from the abnormal functions of the sickle cells, and their inability to flow through the small blood vessels correctly.

Congenital dyserythropoietic anemia (CDA) is a rare blood disorder, similar to the thalassemias. CDA is one of many types of anemia, characterized by ineffective erythropoiesis, and resulting from a decrease in the number of red blood cells (RBCs) in the body and a less than normal quantity of hemoglobin in the blood. CDA may be transmitted by both parents autosomal recessively or dominantly.

Hemoglobin Lepore syndrome is typically an asymptomatic hemoglobinopathy, which is caused by an autosomal recessive genetic mutation. The Hb Lepore variant, consisting of two normal alpha globin chains (HBA) and two delta-beta globin fusion chains which occurs due to a "crossover" between the delta (HBD) and beta globin (HBB) gene loci during meiosis and was first identified in the Lepore family, an Italian-American family, in 1958. There are three varieties of Hb Lepore, Washington, Baltimore and Hollandia. All three varieties show similar electrophoretic and chromatographic properties and hematological findings bear close resemblance to those of the beta-thalassemia trait; a blood disorder that reduces the production of the iron-containing protein hemoglobin which carries oxygen to cells and which may cause anemia.
Neal Stuart Young is an American physician and researcher, chief of the Hematology Branch of the National Institutes of Health (NIH), and Director of the Center for Human Immunology at the NIH in Bethesda, Maryland. He is primarily known for his work in the pathophysiology and treatment of aplastic anemia, and is also known for his contributions to the pathophysiology of parvovirus B19 infection.

Stefan Karlsson is a Professor of Molecular Medicine and Gene Therapy at the Lund Stem Cell Center, in the Department of Laboratory Medicine, Lund University, Sweden. He is recognized for significant contributions to the fields of gene therapy and hematopoietic stem cell biology and in 2009 was awarded the Tobias Prize by The Royal Swedish Academy of Sciences.
Sickle cell-beta thalassemia is an inherited blood disorder. The disease may range in severity from being relatively benign and like sickle cell trait to being similar to sickle cell disease.
Hemoglobin H disease, also called alpha-thalassemia intermedia, is a disease affecting hemoglobin, the oxygen carrying molecule within red blood cells. It is a form of Alpha-thalassemia which most commonly occurs due to deletion of 3 out of 4 of the α-globin genes.

Hemoglobin Hopkins-2 is a mutation of the protein hemoglobin, which is responsible for the transportation of oxygen through the blood from the lungs to the musculature of the body in vertebrates. The specific mutation in Hemoglobin Hopkins-2 results in two abnormal α chains. The mutation is the result of histidine 112 being replaced with aspartic acid in the protein's polypeptide sequence. Additionally, within one of the mutated alpha chains, there are substitutes at 114 and 118, two points on the amino acid chain. This mutation can cause sickle cell anemia.

Samuel Charache was an American hematologist and professor at Johns Hopkins University. He led the research team that discovered the first effective treatment for sickle cell disease, a painful and sometimes fatal blood disorder that mainly affects people of African ancestry.

Swee Lay Thein is a Malaysian haematologist and physician-scientist who is Senior Investigator at the National Institutes of Health. She works on the pathophysiology of haemoglobin disorders including sickle cell disease and thalassemia.

Constance Tom Noguchi is a research physicist, Chief of the Molecular Cell Biology Section, and Dean of the Foundation for Advanced Education in the Sciences (FAES) Graduate School at the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (NIH). Noguchi studies the underlying genetics, metabolism, and treatment of sickle cell disease and of erythropoietin and its effects on metabolism.
Hemoglobin D (HbD) is a variant of hemoglobin, a protein complex that makes up red blood cells. Based on the locations of the original identification, it has been known by several names such as hemoglobin D-Los Angeles, hemoglobin D-Punjab, D-North Carolina, D-Portugal, D-Oak Ridge, and D-Chicago. Hemoglobin D-Los Angeles was the first type identified by Harvey Itano in 1951, and was subsequently discovered that hemoglobin D-Punjab is the most abundant type that is common in the Sikhs of Punjab and of Gujarat.
