The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Suzuki reaction or Suzuki coupling is an organic reaction that uses a palladium complex catalyst to cross-couple a boronic acid to an organohalide. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of noble metal catalysis in organic synthesis. This reaction is sometimes telescoped with the related Miyaura borylation; the combination is the Suzuki–Miyaura reaction. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls.
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
The Ullmann reaction or Ullmann coupling, named after Fritz Ullmann, couples two aryl or alkyl groups with the help of copper. The reaction was first reported by Ullmann and his student Bielecki in 1901. It has been later shown that palladium and nickel can also be effectively used.
The Ullmann condensation or Ullmann-type reaction is the copper-promoted conversion of aryl halides to aryl ethers, aryl thioethers, aryl nitriles, and aryl amines. These reactions are examples of cross-coupling reactions.

Palladium(II) acetate is a chemical compound of palladium described by the formula [Pd(O2CCH3)2]n, abbreviated [Pd(OAc)2]n. It is more reactive than the analogous platinum compound. Depending on the value of n, the compound is soluble in many organic solvents and is commonly used as a catalyst for organic reactions.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.
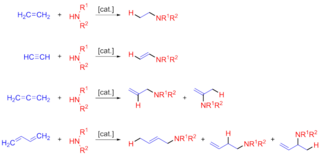
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.

The Liebeskind–Srogl coupling reaction is an organic reaction forming a new carbon–carbon bond from a thioester and a boronic acid using a metal catalyst. It is a cross-coupling reaction. This reaction was invented by and named after Jiri Srogl from the Academy of Sciences, Czech Republic, and Lanny S. Liebeskind from Emory University, Atlanta, Georgia, USA. There are three generations of this reaction, with the first generation shown below. The original transformation used catalytic Pd(0), TFP = tris(2-furyl)phosphine as an additional ligand and stoichiometric CuTC = copper(I) thiophene-2-carboxylate as a co-metal catalyst. The overall reaction scheme is shown below.

DuPhos is a class of organophosphorus compound that are used ligands for asymmetric synthesis. The name DuPhos is derived from (1) the chemical company that sponsored the research leading to this ligand's invention, DuPont and (2) the compound is a diphosphine ligand type. Specifically it is classified as a C2-symmetric ligand, consisting of two phospholanes rings affixed to a benzene ring.
Metal carbon dioxide complexes are coordination complexes that contain carbon dioxide ligands. Aside from the fundamental interest in the coordination chemistry of simple molecules, studies in this field are motivated by the possibility that transition metals might catalyze useful transformations of CO2. This research is relevant both to organic synthesis and to the production of "solar fuels" that would avoid the use of petroleum-based fuels.

PEPPSI is an abbreviation for pyridine-enhanced precatalyst preparation stabilization and initiation. It refers to a family of commercially available palladium catalysts developed around 2005 by Prof. Michael G. Organ and co-workers at York University, which can accelerate various carbon-carbon and carbon-heteroatom bond forming cross-coupling reactions. In comparison to many alternative palladium catalysts, Pd-PEPPSI-type complexes are stable to air and moisture and are relatively easy to synthesize and handle.
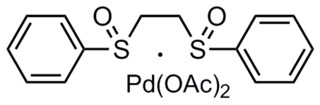
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
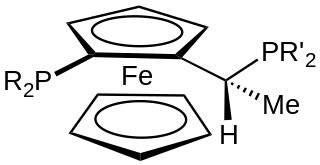
A Josiphos ligand is a type of chiral diphosphine which has been modified to be substrate-specific; they are widely used for enantioselective synthesis. They are widely used in asymmetric catalysis.
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.

Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Colorless solids, PHOX ligands feature a tertiary phosphine group, often diphenyl, and an oxazoline ligand in the ortho position. The oxazoline, which carries the stereogenic center, coordinates through nitrogen, the result being that PHOX ligands are P,N-chelating ligands. Most phosphine ligands used in asymmetric catalysis are diphosphines, so the PHOX ligands are distinctive. Some evidence exists that PHOX ligands are hemilabile.

Alkene carboamination is the simultaneous formation of C–N and C–C bonds across an alkene. This method represents a powerful strategy to build molecular complexity with up to two stereocenters in a single operation. Generally, there are four categories of reaction modes for alkene carboamination. The first class is cyclization reactions, which will form a N-heterocycle as a result. The second class has been well established in the last decade. Alkene substrates with a tethered nitrogen nucleophile have been used in these transformations to promote intramolecular aminocyclization. While intermolecular carboamination is extremely hard, people have developed a strategy to combine the nitrogen and carbon part, which is known as the third class. The most general carboamination, which takes three individual parts and couples them together is still underdeveloped.
Dialkylbiaryl phosphine ligands are phosphine ligands that are used in homogeneous catalysis. They have proved useful in Buchwald-Hartwig amination and etherification reactions as well as Negishi cross-coupling, Suzuki-Miyaura cross-coupling, and related reactions. In addition to these Pd-based processes, their use has also been extended to transformations catalyzed by nickel, gold, silver, copper, rhodium, and ruthenium, among other transition metals.

The nitro-Mannich reaction is the nucleophilic addition of a nitroalkane to an imine, resulting in the formation of a beta-nitroamine. With the reaction involving the addition of an acidic carbon nucleophile to a carbon-heteroatom double bond, the nitro-Mannich reaction is related to some of the most fundamental carbon-carbon bond forming reactions in organic chemistry, including the aldol reaction, Henry reaction and Mannich reaction.








