The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.
In chemistry, transfer hydrogenation is a chemical reaction involving the addition of hydrogen to a compound from a source other than molecular H2. It is applied in laboratory and industrial organic synthesis to saturate organic compounds and reduce ketones to alcohols, and imines to amines. It avoids the need for high-pressure molecular H2 used in conventional hydrogenation. Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis. It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively. Often, the donor molecules also function as solvents for the reaction. A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
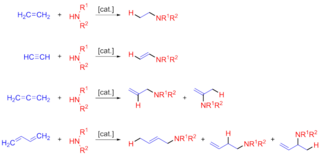
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being awarded one half of the 2001 Nobel Prize in Chemistry.

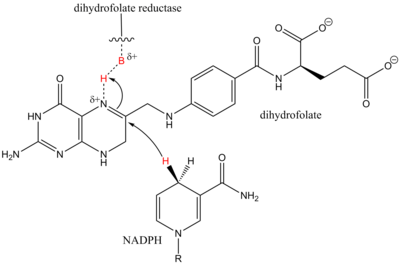
In chemistry, the hydrogenation of carbon–nitrogen double bonds is the addition of the elements of dihydrogen (H2) across a carbon–nitrogen double bond, forming amines or amine derivatives. Although a variety of general methods have been developed for the enantioselective hydrogenation of ketones, methods for the hydrogenation of carbon–nitrogen double bonds are less general. Hydrogenation of imines is complicated by both syn/anti isomerization and tautomerization to enamines, which may be hydrogenated with low enantioselectivity in the presence of a chiral catalyst. Additionally, the substituent attached to nitrogen affects both the reactivity and spatial properties of the imine, complicating the development of a general catalyst system for imine hydrogenation. Despite these challenges, methods have been developed that address particular substrate classes, such as N-aryl, N-alkyl, and endocyclic imines.

Hydrogen auto-transfer, also known as borrowing hydrogen, is the activation of a chemical reaction by temporary transfer of two hydrogen atoms from the reactant to a catalyst and return of those hydrogen atoms back to a reaction intermediate to form the final product. Two major classes of borrowing hydrogen reactions exist: (a) those that result in hydroxyl substitution, and (b) those that result in carbonyl addition. In the former case, alcohol dehydrogenation generates a transient carbonyl compound that is subject to condensation followed by the return of hydrogen. In the latter case, alcohol dehydrogenation is followed by reductive generation of a nucleophile, which triggers carbonyl addition. As borrowing hydrogen processes avoid manipulations otherwise required for discrete alcohol oxidation and the use of stoichiometric organometallic reagents, they typically display high levels of atom-economy and, hence, are viewed as examples of Green chemistry.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
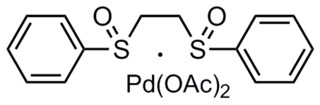
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.

Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Their complexes are particularly effective at generating single enatiomers in reactions involving highly symmetric transition states, such as allylic substitutions, which are typically difficult to perform stereoselectively. The ligands are bidentate and have been shown to be hemilabile with the softer P‑donor being more firmly bound than the harder N‑donor.
In organic chemistry, the Keck asymmetric allylation is a chemical reaction that involves the nucleophilic addition of an allyl group to an aldehyde. The catalyst is a chiral complex that contains titanium as a Lewis acid. The chirality of the catalyst induces a stereoselective addition, so the secondary alcohol of the product has a predictable absolute stereochemistry based on the choice of catalyst. This name reaction is named for Gary Keck.
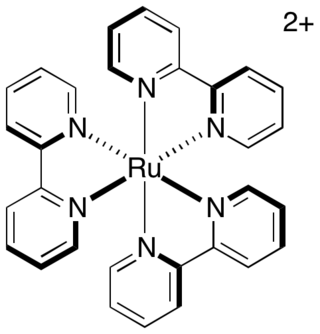
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.

The A3 coupling (also known as A3 coupling reaction or the aldehyde-alkyne-amine reaction), coined by Prof. Chao-Jun Li of McGill University, is a type of multicomponent reaction involving an aldehyde, an alkyne and an amine which react to give a propargylamine.

The Krische allylation involves the enantioselective iridium-catalyzed addition of an allyl group to an aldehyde or an alcohol, resulting in the formation of a secondary homoallylic alcohol. The mechanism of the Krische allylation involves primary alcohol dehydrogenation or, when using aldehyde reactants, hydrogen transfer from 2-propanol. Unlike other allylation methods, the Krische allylation avoids the use of preformed allyl metal reagents and enables the direct conversion of primary alcohols to secondary homoallylic alcohols.
In homogeneous catalysis, C2-symmetric ligands refer to ligands that lack mirror symmetry but have C2 symmetry. Such ligands are usually bidentate and are valuable in catalysis. The C2 symmetry of ligands limits the number of possible reaction pathways and thereby increases enantioselectivity, relative to asymmetrical analogues. C2-symmetric ligands are a subset of chiral ligands. Chiral ligands, including C2-symmetric ligands, combine with metals or other groups to form chiral catalysts. These catalysts engage in enantioselective chemical synthesis, in which chirality in the catalyst yields chirality in the reaction product.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.
Copper-catalyzed allylic substitutions are chemical reactions with unique regioselectivity compared to other transition-metal-catalyzed allylic substitutions such as the Tsuji-Trost reaction. They involve copper catalysts and "hard" carbon nucleophiles. The mechanism of copper-catalyzed allylic substitutions involves the coordination of copper to the olefin, oxidative addition and reductive elimination. Enantioselective versions of these reactions have been used in the synthesis of complex molecules, such as (R)-(-)-sporochnol and (S)-(-)-Zearalenone.