Histocompatibility, or tissue compatibility, is the property of having the same, or sufficiently similar, alleles of a set of genes called human leukocyte antigens (HLA), or major histocompatibility complex (MHC). Each individual expresses many unique HLA proteins on the surface of their cells, which signal to the immune system whether a cell is part of the self or an invading organism. T cells recognize foreign HLA molecules and trigger an immune response to destroy the foreign cells. Histocompatibility testing is most relevant for topics related to whole organ, tissue, or stem cell transplants, where the similarity or difference between the donor's HLA alleles and the recipient's triggers the immune system to reject the transplant. The wide variety of potential HLA alleles lead to unique combinations in individuals and make matching difficult.

The major histocompatibility complex (MHC) is a large locus on vertebrate DNA containing a set of closely linked polymorphic genes that code for cell surface proteins essential for the adaptive immune system. These cell surface proteins are called MHC molecules.

The human leukocyte antigen (HLA) system is a complex of genes on chromosome 6 in humans that encode cell-surface proteins responsible for regulation of the immune system. The HLA system is also known as the human version of the major histocompatibility complex (MHC) found in many animals.

Transplant rejection occurs when transplanted tissue is rejected by the recipient's immune system, which destroys the transplanted tissue. Transplant rejection can be lessened by determining the molecular similitude between donor and recipient and by use of immunosuppressant drugs after transplant.
Alloimmunity is an immune response to nonself antigens from members of the same species, which are called alloantigens or isoantigens. Two major types of alloantigens are blood group antigens and histocompatibility antigens. In alloimmunity, the body creates antibodies against the alloantigens, attacking transfused blood, allotransplanted tissue, and even the fetus in some cases. Alloimmune (isoimmune) response results in graft rejection, which is manifested as deterioration or complete loss of graft function. In contrast, autoimmunity is an immune response to the self's own antigens. Alloimmunization (isoimmunization) is the process of becoming alloimmune, that is, developing the relevant antibodies for the first time.

Jean-Baptiste-Gabriel-Joachim Dausset was a French immunologist born in Toulouse, France. Dausset received the Nobel Prize in Physiology or Medicine in 1980 along with Baruj Benacerraf and George Davis Snell for their discovery and characterisation of the genes making the major histocompatibility complex. Using the money from his Nobel Prize and a grant from the French Television, Dausset founded the Human Polymorphism Study Center (CEPH) in 1984, which was later renamed the Foundation Jean Dausset-CEPH in his honour. He married Rose Mayoral in 1963, with whom he had two children, Henri and Irène. Jean Dausset died on June 6, 2009, in Majorca, Spain, at the age of 92.

Jan Klein was a Czech–American immunologist.

HLA-DQ (DQ) is a cell surface receptor protein found on antigen-presenting cells. It is an αβ heterodimer of type MHC class II. The α and β chains are encoded by two loci, HLA-DQA1 and HLA-DQB1, that are adjacent to each other on chromosome band 6p21.3. Both α-chain and β-chain vary greatly. A person often produces two α-chain and two β-chain variants and thus 4 isoforms of DQ. The DQ loci are in close genetic linkage to HLA-DR, and less closely linked to HLA-DP, HLA-A, HLA-B and HLA-C.
HLA-DP is a protein/peptide-antigen receptor and graft-versus-host disease antigen that is composed of 2 subunits, DPα and DPβ. DPα and DPβ are encoded by two loci, HLA-DPA1 and HLA-DPB1, that are found in the MHC Class II region in the Human Leukocyte Antigen complex on human chromosome 6 . Less is known about HLA-DP relative to HLA-DQ and HLA-DR but the sequencing of DP types and determination of more frequent haplotypes has progressed greatly within the last few years.
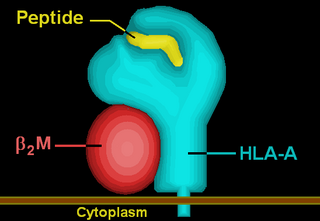
HLA-A is a group of human leukocyte antigens (HLA) that are encoded by the HLA-A locus, which is located at human chromosome 6p21.3. HLA is a major histocompatibility complex (MHC) antigen specific to humans. HLA-A is one of three major types of human MHC class I transmembrane proteins. The others are HLA-B and HLA-C. The protein is a heterodimer, and is composed of a heavy α chain and smaller β chain. The α chain is encoded by a variant HLA-A gene, and the β chain (β2-microglobulin) is an invariant β2 microglobulin molecule. The β2 microglobulin protein is encoded by the B2M gene, which is located at chromosome 15q21.1 in humans.
Tissue typing is a procedure in which the tissues of a prospective donor and recipient are tested for compatibility prior to transplantation. Mismatched donor and recipient tissues can lead to rejection of the tissues. There are multiple methods of tissue typing.

Minor histocompatibility antigen are peptides presented on the cellular surface of donated organs that are known to give an immunological response in some organ transplants. They cause problems of rejection less frequently than those of the major histocompatibility complex (MHC). Minor histocompatibility antigens (MiHAs) are diverse, short segments of proteins and are referred to as peptides. These peptides are normally around 9-12 amino acids in length and are bound to both the major histocompatibility complex (MHC) class I and class II proteins. Peptide sequences can differ among individuals and these differences arise from SNPs in the coding region of genes, gene deletions, frameshift mutations, or insertions. About a third of the characterized MiHAs come from the Y chromosome. Prior to becoming a short peptide sequence, the proteins expressed by these polymorphic or diverse genes need to be digested in the proteasome into shorter peptides. These endogenous or self peptides are then transported into the endoplasmic reticulum with a peptide transporter pump called TAP where they encounter and bind to the MHC class I molecule. This contrasts with MHC class II molecules's antigens which are peptides derived from phagocytosis/endocytosis and molecular degradation of non-self entities' proteins, usually by antigen-presenting cells. MiHA antigens are either ubiquitously expressed in most tissue like skin and intestines or restrictively expressed in the immune cells.
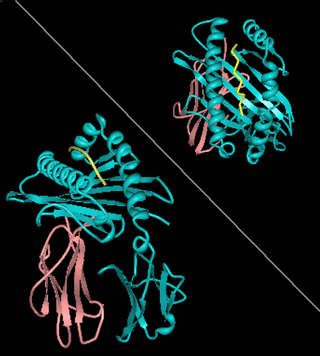
HLA-A1 (A1) is a human leukocyte antigen serotype within HLA-A "A" serotype group. The serotype is determined by the antibody recognition of α1 subset of HLA-A α-chains. For A1, the alpha "A" chain are encoded by the HLA-A*01 allele group and the β-chain are encoded by B2M locus. This group currently is dominated by A*01:01. A1 and A*01 are almost synonymous in meaning. A1 is more common in Europe than elsewhere, it is part of a long haplotype that appears to have been frequent in the ancient peoples of Northwestern Europe. A1 is a frequent component of the AH8.1 haplotype. A1 serotype positivity is roughly linked to a large number of inflammatory diseases and conditions believed to have immune system involvement. Because of its linkage within the AH8.1 haplotype many studies showed association with A1 or A1,B8 only later to show the association drift toward the class II region gene alleles, DR3 and DQ2.5. While it is not clear what role A1 has in infectious disease, some linkage with infection rates in HIV remain associated within the A1 region of the haplotype.

HLA-A*02 (A*02) is a human leukocyte antigen serotype within the HLA-A serotype group. The serotype is determined by the antibody recognition of the α2 domain of the HLA-A α-chain. For A*02, the α chain is encoded by the HLA-A*02 gene and the β chain is encoded by the B2M locus. In 2010 the World Health Organization Naming Committee for Factors of the HLA System revised the nomenclature for HLAs. Before this revision, HLA-A*02 was also referred to as HLA-A2, HLA-A02, and HLA-A*2.
HLA-A3 (A3) is a human leukocyte antigen serotype within HLA-A serotype group. The serotype is determined by the antibody recognition of α3 subset of HLA-A α-chains. For A3, the alpha, "A", chain are encoded by the HLA-A*03 allele group and the β-chain are encoded by B2M locus. This group currently is dominated by A*03:01. A3 and A*03 are almost synonymous in meaning. A3 is more common in Europe, it is part of the longest known multigene haplotype, A3~B7~DR15~DQ6.
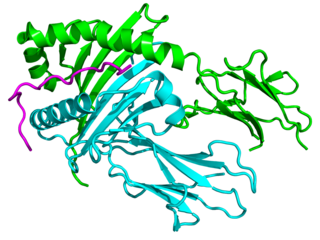
Major histocompatibility complex, class II, DQ alpha 1, also known as HLA-DQA1, is a human gene present on short arm of chromosome 6 (6p21.3) and also denotes the genetic locus which contains this gene. The protein encoded by this gene is one of two proteins that are required to form the DQ heterodimer, a cell surface receptor essential to the function of the immune system.
HLA A1-B8-DR3-DQ2 haplotype is a multigene haplotype that covers a majority of the human major histocompatibility complex on chromosome 6. A multigene haplotype is set of inherited alleles covering several genes, or gene-alleles; common multigene haplotypes are generally the result of descent by common ancestry. Chromosomal recombination fragments multigene haplotypes as the distance to that ancestor increases in number of generations.
HLA A1-B8 is a multigene haplotype that covers the MHC Class I region of the human major histocompatibility complex on chromosome 6. A multigene haplotype is set of inherited alleles covering several genes, or gene-alleles; common multigene haplotypes are generally the result of identity by descent from a common ancestor. Chromosomal recombination fragments multigene haplotypes as the distance to that ancestor increases in number of generations.
HLA-Cw7 (Cw7) is a human leukocyte antigen serotype within HLA-C serotype group. Cw7 is determined by the antibody recognition of α7 subset of HLA-Cw α-chains. For the serotype Cw7, the alpha chain are encoded by the HLA-Cw*07 allele group and the β-chain are encoded by B2M locus. Cw7 and Cw*07 are almost synonymous in meaning. Cw7 is more common in West Africa to Ireland. Cw7 in Europe is part of the AH8.1 and HLA B7-DR15-DQ6 haplotypes. The class I region of these supertype is HLA A1-B8 haplotype, HLA A3-B7, HLA-A2-B7 and HLA A24-B7.
HLA B7-DR15-DQ6 is a multigene haplotype that covers a majority of the human major histocompatibility complex on chromosome 6. A multigene haplotype is set of inherited alleles covering several genes, or gene-alleles, common multigene haplotypes are generally the result of descent by common ancestry. Chromosomal recombination fragments multigene haplotypes as the distance to that ancestor increases in number of generations.







