Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
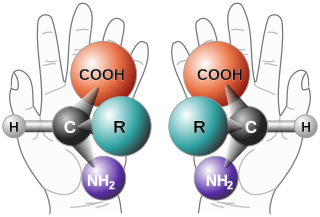
Enantioselective synthesis, also called asymmetric synthesis, is a form of chemical synthesis. It is defined by IUPAC as "a chemical reaction in which one or more new elements of chirality are formed in a substrate molecule and which produces the stereoisomeric products in unequal amounts."
In chemistry, stereoselectivity is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. The selectivity arises from differences in steric and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite. Both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
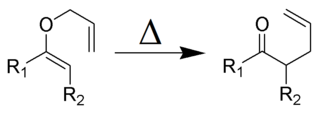
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl.

A chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal. The metal is often used as a catalyst, with some other stoichiometric oxidant present. In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.
Longifolene is the common chemical name of a naturally occurring, oily liquid hydrocarbon found primarily in the high-boiling fraction of certain pine resins. The name is derived from that of a pine species from which the compound was isolated, Pinus longifolia

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. This enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes. Kinetic resolution is a concept in organic chemistry and can be used for the preparation of chiral molecules in organic synthesis. Kinetic resolution reactions utilizing purely synthetic reagents and catalysts are much less common than the use of enzymatic kinetic resolution in application towards organic synthesis, although a number of useful synthetic techniques have been developed in the past 30 years.
A chiral derivatizing agent (CDA) also known as a chiral resolving reagent, is a chiral auxiliary used to convert a mixture of enantiomers into diastereomers in order to analyze the quantities of each enantiomer present within the mix. Analysis can be conducted by spectroscopy or by chromatography. The use of chiral derivatizing agents has declined with the popularization of chiral HPLC. Besides analysis, chiral derivatization is also used for chiral resolution, the actual physical separation of the enantiomers.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
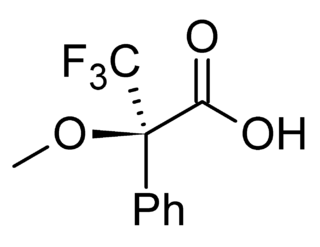
Mosher's acid, or α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) is a carboxylic acid which was first used by Harry Stone Mosher as a chiral derivatizing agent. It is a chiral molecule, consisting of R and S enantiomers.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
In organic chemistry, enone–alkene cycloadditions are a version of the [2+2] cycloaddition This reaction involves an enone and alkene as substrates. Although the concerted photochemical [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.
The imine Diels–Alder reaction involves the transformation of all-carbon dienes and imine dienophiles into tetrahydropyridines.

Torquoselectivity is a special kind of stereoselectivity observed in electrocyclic reactions in organic chemistry, defined as "the preference for inward or outward rotation of substituents in conrotatory or disrotatory electrocyclic reactions." Torquoselectivity is not to be confused with the normal diastereoselectivity seen in pericyclic reactions, as it represents a further level of selectivity beyond the Woodward-Hoffman rules. The name derives from the idea that the substituents in an electrocyclization appear to rotate over the course of the reaction, and thus selection of a single product is equivalent to selection of one direction of rotation. The concept was originally developed by Kendall N. Houk.

Camphorsultam, also known as bornanesultam, is a crystalline solid primarily used as a chiral auxiliary in the synthesis of other chemicals with a specific desired stereoselectivity. Camphorsultam is commercially available in both enantiomers of its exo forms: (1R)-(+)-2,10-camphorsultam and (1S)-(−)-2,10-camphorsultam.
The [4+4] Photocycloaddition is a cycloaddition reaction in which two unsaturated molecules connect via four atoms from each molecule to create an eight-membered ring. As a photochemical reaction, it is promoted by some form of light, as opposed to a thermal process.

Ugi’s amine is a chemical compound named for the chemist who first reported its synthesis in 1970, Ivar Ugi. It is a ferrocene derivative. Since its first report, Ugi’s amine has found extensive use as the synthetic precursor to a large number of metal ligands that bear planar chirality. These ligands have since found extensive use in a variety of catalytic reactions. The compound may exist in either the 1S or 1R isomer, both of which have synthetic utility and are commercially available. Most notably, it is the synthetic precursor to the Josiphos class of ligands.


