
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Cranial nerves are the nerves that emerge directly from the brain, of which there are conventionally considered twelve pairs. Cranial nerves relay information between the brain and parts of the body, primarily to and from regions of the head and neck, including the special senses of vision, taste, smell, and hearing.

Bell's palsy is a type of facial paralysis that results in a temporary inability to control the facial muscles on the affected side of the face. In most cases, the weakness is temporary and significantly improves over weeks. Symptoms can vary from mild to severe. They may include muscle twitching, weakness, or total loss of the ability to move one or, in rare cases, both sides of the face. Other symptoms include drooping of the eyebrow, a change in taste, and pain around the ear. Typically symptoms come on over 48 hours. Bell's palsy can trigger an increased sensitivity to sound known as hyperacusis.
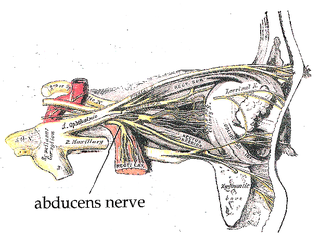
The abducens nerve or abducent nerve, also known as the sixth cranial nerve, cranial nerve VI, or simply CN VI, is a cranial nerve in humans and various other animals that controls the movement of the lateral rectus muscle, one of the extraocular muscles responsible for outward gaze. It is a somatic efferent nerve.

The hypoglossal nerve, also known as the twelfth cranial nerve, cranial nerve XII, or simply CN XII, is a cranial nerve that innervates all the extrinsic and intrinsic muscles of the tongue except for the palatoglossus, which is innervated by the vagus nerve.

The somatic nervous system (SNS), also known as voluntary nervous system, is a part of the peripheral nervous system (PNS) that links brain and spinal cord to skeletal muscles under conscious control, as well as to sensory receptors in the skin. The other part complementary to the somatic nervous system is the autonomic nervous system (ANS).

The pyramidal tracts include both the corticobulbar tract and the corticospinal tract. These are aggregations of efferent nerve fibers from the upper motor neurons that travel from the cerebral cortex and terminate either in the brainstem (corticobulbar) or spinal cord (corticospinal) and are involved in the control of motor functions of the body.

Facial nerve paralysis is a common problem that involves the paralysis of any structures innervated by the facial nerve. The pathway of the facial nerve is long and relatively convoluted, so there are a number of causes that may result in facial nerve paralysis. The most common is Bell's palsy, a disease of unknown cause that may only be diagnosed by exclusion of identifiable serious causes.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.

Fazio–Londe disease (FLD), also called progressive bulbar palsy of childhood, is a very rare inherited motor neuron disease of children and young adults and is characterized by progressive paralysis of muscles innervated by cranial nerves. FLD, along with Brown–Vialetto–Van Laere syndrome (BVVL), are the two forms of infantile progressive bulbar palsy, a type of progressive bulbar palsy in children.
Progressive bulbar palsy (PBP) is a medical condition. It belongs to a group of disorders known as motor neuron diseases. PBP is a disease that attacks the nerves supplying the bulbar muscles. These disorders are characterized by the degeneration of motor neurons in the cerebral cortex, spinal cord, brain stem, and pyramidal tracts. This specifically involves the glossopharyngeal nerve (IX), vagus nerve (X), and hypoglossal nerve (XII).
Pseudobulbar palsy is a medical condition characterized by the inability to control facial movements and caused by a variety of neurological disorders. Patients experience difficulty chewing and swallowing, have increased reflexes and spasticity in tongue and the bulbar region, and demonstrate slurred speech, sometimes also demonstrating uncontrolled emotional outbursts.
The cerebellopontine angle syndrome is a distinct neurological syndrome of deficits that can arise due to the closeness of the cerebellopontine angle to specific cranial nerves. Indications include unilateral hearing loss (85%), speech impediments, disequilibrium, tremors or other loss of motor control. The cerebellopontine angle cistern is a subarachnoid cistern formed by the cerebellopontine angle that lies between the cerebellum and the pons. It is filled with cerebrospinal fluid and is a common site for the growth of acoustic neuromas or schwannomas.

Cranial nerve disease is an impaired functioning of one of the twelve cranial nerves. Although it could theoretically be considered a mononeuropathy, it is not considered as such under MeSH.
Brown-Vialetto-Van-Laere syndrome (BVVL), sometimes known as Brown's syndrome, is a rare degenerative disorder often initially characterized by progressive sensorineural deafness.

Hereditary neuropathy with liability to pressure palsy (HNPP) is a peripheral neuropathy, a condition that affects the nerves. Pressure on the nerves can cause tingling sensations, numbness, pain, weakness, muscle atrophy and even paralysis of the affected area. In normal individuals, these symptoms disappear quickly, but in sufferers of HNPP even a short period of pressure can cause the symptoms to occur. Palsies can last from minutes or days to weeks or even months.
Superficial hemosiderosis of the central nervous system is a disease of the brain resulting from chronic iron deposition in neuronal tissues associated with cerebrospinal fluid. This occurs via the deposition of hemosiderin in neuronal tissue, and is associated with neuronal loss, gliosis, and demyelination of neuronal cells. This disease was first discovered in 1908 by R.C. Hamill after performing an autopsy. Detection of this disease was largely post-mortem until the advent of MRI technology, which made diagnosis far easier. Superficial siderosis is largely considered a rare disease, with less than 270 total reported cases in scientific literature as of 2006, and affects people of a wide range of ages with men being approximately three times more frequently affected than women. The number of reported cases of superficial siderosis has increased with advances in MRI technology, but it remains a rare disease.
Alternating hemiplegia is a form of hemiplegia that has an ipsilateral cranial nerve palsies and contralateral hemiplegia or hemiparesis of extremities of the body. The disorder is characterized by recurrent episodes of paralysis on one side of the body. There are multiple forms of alternating hemiplegia, Weber's syndrome, middle alternating hemiplegia, and inferior alternating hemiplegia. This type of syndrome can result from a unilateral lesion in the brainstem affecting both upper motor neurons and lower motor neurons. The muscles that would receive signals from these damaged upper motor neurons result in spastic paralysis. With a lesion in the brainstem, this affects the majority of limb and trunk muscles on the contralateral side due to the upper motor neurons decussation after the brainstem. The cranial nerves and cranial nerve nuclei are also located in the brainstem making them susceptible to damage from a brainstem lesion. Cranial nerves III (Oculomotor), VI (Abducens), and XII (Hypoglossal) are most often associated with this syndrome given their close proximity with the pyramidal tract, the location which upper motor neurons are in on their way to the spinal cord. Damages to these structures produce the ipsilateral presentation of paralysis or palsy due to the lack of cranial nerve decussation before innervating their target muscles. The paralysis may be brief or it may last for several days, many times the episodes will resolve after sleep. Some common symptoms of alternating hemiplegia are mental impairment, gait and balance difficulties, excessive sweating and changes in body temperature.

Hirayama disease, also known as monomelic amyotrophy (MMA), is a rare motor neuron disease first described in 1959 in Japan. Its symptoms usually appear about two years after adolescent growth spurt and is significantly more common in males, with an average age of onset between 15 and 25 years. Hirayama disease is reported most frequently in Asia but has a global distribution. It is typically marked by insidious onset of muscle atrophy of an upper limb, which plateaus after two to five years from which it neither improves nor worsens. There is no pain or sensory loss. It is not believed to be hereditary.
Facial onset sensory and motor neuronopathy, often abbreviated FOSMN, is a rare disorder of the nervous system in which sensory and motor nerves of the face and limbs progressively degenerate over a period of months to years. This degenerative process, the cause of which is unknown, eventually results in sensory and motor symptoms — the former consisting mainly of paresthesia followed by numbness, and the latter in muscle weakness, atrophy, and eventual paralysis. FOSM is characterized by sensory and motor loss beginning in the face and spreading to involve an increasingly larger area including the scalp, upper arms and trunk. The muscles or respiration and swallowing are commonly affected. In many ways, it is reminiscent of the much better known condition amyotrophic lateral sclerosis, with which it is closely related. There is no cure; treatment is supportive. Life expectancy may be shortened by respiratory complications arising from weakness of the muscles that aid breathing and swallowing. It was first described in four patients by Vucic and colleagues working at the Massachusetts General Hospital in the United States; subsequent reports from the United Kingdom, Europe and Asia point to a global incidence of the disease. It is thought to be exceptionally rare, with only approximately 100 individuals described to date in the medical literature.

