Related Research Articles
Vibronic coupling in a molecule involves the interaction between electronic and nuclear vibrational motion. The term "vibronic" originates from the combination of the terms "vibrational" and "electronic", denoting the idea that in a molecule, vibrational and electronic interactions are interrelated and influence each other. The magnitude of vibronic coupling reflects the degree of such interrelation.
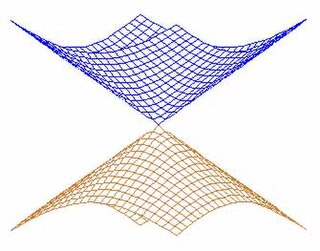
In quantum chemistry, a conical intersection of two or more potential energy surfaces is the set of molecular geometry points where the potential energy surfaces are degenerate (intersect) and the non-adiabatic couplings between these states are non-vanishing. In the vicinity of conical intersections, the Born–Oppenheimer approximation breaks down and the coupling between electronic and nuclear motion becomes important, allowing non-adiabatic processes to take place. The location and characterization of conical intersections are therefore essential to the understanding of a wide range of important phenomena governed by non-adiabatic events, such as photoisomerization, photosynthesis, vision and the photostability of DNA. The conical intersection involving the ground electronic state potential energy surface of the C6H3F3+ molecular ion is discussed in connection with the Jahn–Teller effect in Section 13.4.2 on pages 380-388 of the textbook by Bunker and Jensen.

Photosensitizers are light absorbers that alter the course of a photochemical reaction. They usually are catalysts. They can function by many mechanisms, sometimes they donate an electron to the substrate, sometimes they abstract a hydrogen atom from the substrate. At the end of this process, the photosensitizer returns to its ground state, where it remains chemically intact, poised to absorb more light. One branch of chemistry which frequently utilizes photosensitizers is polymer chemistry, using photosensitizers in reactions such as photopolymerization, photocrosslinking, and photodegradation. Photosensitizers are also used to generate prolonged excited electronic states in organic molecules with uses in photocatalysis, photon upconversion and photodynamic therapy. Generally, photosensitizers absorb electromagnetic radiation consisting of infrared radiation, visible light radiation, and ultraviolet radiation and transfer absorbed energy into neighboring molecules. This absorption of light is made possible by photosensitizers' large de-localized π-systems, which lowers the energy of HOMO and LUMO orbitals to promote photoexcitation. While many photosensitizers are organic or organometallic compounds, there are also examples of using semiconductor quantum dots as photosensitizers.
Semi-empirical quantum chemistry methods are based on the Hartree–Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree–Fock method without the approximations is too expensive. The use of empirical parameters appears to allow some inclusion of electron correlation effects into the methods.
Todd J. Martínez is a David Mulvane Ehrsam and Edward Curtis Franklin Professor of Chemistry at Stanford University and a Professor of Photon Science at the SLAC National Accelerator Laboratory.
The ab initio multiple spawning, or AIMS, method is a time-dependent formulation of quantum chemistry.

Anna Igorevna Krylov is the USC Associates Chair in Natural Sciences and Professor of Chemistry at the University of Southern California (USC). Working in the field of theoretical and computational quantum chemistry, she is the inventor of the spin-flip method. Krylov is the president of Q-Chem, Inc. and an elected member of the International Academy of Quantum Molecular Science, the Academia Europaea, and the American Academy of Sciences and Letters.
In organic chemistry, the di-π-methane rearrangement is the photochemical rearrangement of a molecule that contains two π-systems separated by a saturated carbon atom. In the aliphatic case, this molecules is a 1,4-diene; in the aromatic case, an allyl-substituted arene. The reaction forms (respectively) an ene- or aryl-substituted cyclopropane. Formally, it amounts to a 1,2 shift of one ene group or the aryl group, followed by bond formation between the lateral carbons of the non-migrating moiety:
Quantum biology is the study of applications of quantum mechanics and theoretical chemistry to aspects of biology that cannot be accurately described by the classical laws of physics. An understanding of fundamental quantum interactions is important because they determine the properties of the next level of organization in biological systems.

Photomagnetism is the effect in which a material acquires its ferromagnetic properties in response to light. The current model for this phenomenon is a light-induced electron transfer, accompanied by the reversal of the spin direction of an electron. This leads to an increase in spin concentration, causing the magnetic transition. Currently the effect is only observed to persist at very low temperature. But at temperatures such as 5K, the effect may persist for several days.

Photon upconversion (UC) is a process in which the sequential absorption of two or more photons leads to the emission of light at shorter wavelength than the excitation wavelength. It is an anti-Stokes type emission. An example is the conversion of infrared light to visible light. Upconversion can take place in both organic and inorganic materials, through a number of different mechanisms. Organic molecules that can achieve photon upconversion through triplet-triplet annihilation are typically polycyclic aromatic hydrocarbons (PAHs). Inorganic materials capable of photon upconversion often contain ions of d-block or f-block elements. Examples of these ions are Ln3+, Ti2+, Ni2+, Mo3+, Re4+, Os4+, and so on.
Surface hopping is a mixed quantum-classical technique that incorporates quantum mechanical effects into molecular dynamics simulations. Traditional molecular dynamics assume the Born-Oppenheimer approximation, where the lighter electrons adjust instantaneously to the motion of the nuclei. Though the Born-Oppenheimer approximation is applicable to a wide range of problems, there are several applications, such as photoexcited dynamics, electron transfer, and surface chemistry where this approximation falls apart. Surface hopping partially incorporates the non-adiabatic effects by including excited adiabatic surfaces in the calculations, and allowing for 'hops' between these surfaces, subject to certain criteria.

Newton-X is a general program for molecular dynamics simulations beyond the Born-Oppenheimer approximation. It has been primarily used for simulations of ultrafast processes in photoexcited molecules. It has also been used for simulation of band envelops of absorption and emission spectra.
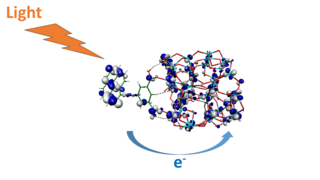
Quantum photoelectrochemistry is the investigation of the quantum mechanical nature of photoelectrochemistry, the subfield of study within physical chemistry concerned with the interaction of light with electrochemical systems, typically through the application of quantum chemical calculations. Quantum photoelectrochemistry provides an expansion of quantum electrochemistry to processes involving also the interaction with light (photons). It therefore also includes essential elements of photochemistry. Key aspects of quantum photoelectrochemistry are calculations of optical excitations, photoinduced electron and energy transfer processes, excited state evolution, as well as interfacial charge separation and charge transport in nanoscale energy conversion systems.

Oleg V. Prezhdo is a Ukrainian–American physical chemist whose research focuses on non-adiabatic molecular dynamics and time-dependent density functional theory (TDDFT). His research interests range from fundamental aspects of semi-classical and quantum-classical physics to excitation dynamics in condensed matter and biological systems. His research group focuses on the development of new theoretical models and computational tools aimed at understanding chemical reactivity and energy transfer at a molecular level in complex condensed phase environment. Since 2014, he is a professor of chemistry and of physics & astronomy at the University of Southern California.
Formamide-based prebiotic chemistry is a reconstruction of the beginnings of life on Earth, assuming that formamide could accumulate in sufficiently high amounts to serve as the building block and reaction medium for the synthesis of the first biogenic molecules.

Hans Lischka is an Austrian computational theoretical chemist specialized on development and application of multireference methods for the study of molecular excited states. He is the main developer of the software package Columbus for ab initio multireference calculations and co-developer of the Newton-X program.
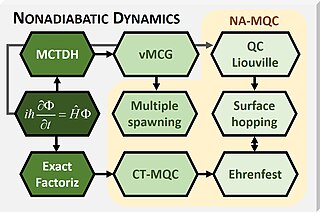
Mixed quantum-classical (MQC) dynamics is a class of computational theoretical chemistry methods tailored to simulate non-adiabatic (NA) processes in molecular and supramolecular chemistry. Such methods are characterized by:
- Propagation of nuclear dynamics through classical trajectories;
- Propagation of the electrons through quantum methods;
- A feedback algorithm between the electronic and nuclear subsystems to recover nonadiabatic information.
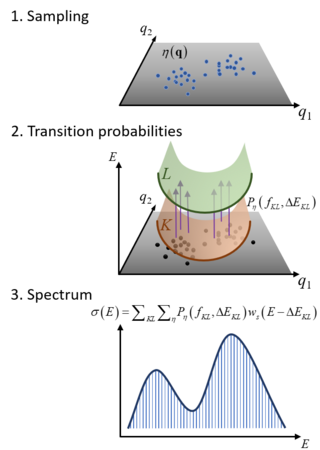
The Nuclear Ensemble Approach (NEA) is a general method for simulations of diverse types of molecular spectra. It works by sampling an ensemble of molecular conformations in the source state, computing the transition probabilities to the target states for each of these geometries, and performing a sum over all these transitions convoluted with shape function. The result is an incoherent spectrum containing absolute band shapes through inhomogeneous broadening.

Dimitra Markovitsi is a Greek-French photochemist. She is currently an Emeritus Research Director at the French National Center for Scientific Research (CNRS). She pioneered studies on the electronically excited states of liquid crystals and made significant advances to the understanding of processes triggered in DNA upon absorption of UV radiation. The two facets of her work have been the subject of a recent Marie Skodowska Curie European training network entitled "Light DyNAmics - DNA as a training platform for photodynamic processes in soft materials."
References
- ↑ Crespo-Otero, Rachel; Barbatti, Mario (2018-08-08). "Recent Advances and Perspectives on Nonadiabatic Mixed Quantum–Classical Dynamics" (PDF). Chemical Reviews. 118 (15): 7026–7068. doi:10.1021/acs.chemrev.7b00577. ISSN 0009-2665. PMID 29767966.
- ↑ "European Academy of Sciences - Mario Barbatti". www.eurasc.org.
- ↑ "Les membres - Institut Universitaire de France". www.iufrance.fr. Retrieved 1 July 2021.
- ↑ "Mario Barbatti - Google Scholar Citations". scholar.google.de. Retrieved 2019-12-29.
- ↑ Barbatti, Mario; Ruckenbauer, Matthias; Plasser, Felix; Pittner, Jiri; Granucci, Giovanni; Persico, Maurizio; Lischka, Hans (2014). "Newton-X: a surface-hopping program for nonadiabatic molecular dynamics". WIREs Computational Molecular Science. 4 (1): 26–33. doi:10.1002/wcms.1158. ISSN 1759-0884. S2CID 60777813.
- ↑ Barbatti, Mario; Aquino, Adélia J. A.; Szymczak, Jaroslaw J.; Nachtigallová, Dana; Hobza, Pavel; Lischka, Hans (2010-12-14). "Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases". Proceedings of the National Academy of Sciences. 107 (50): 21453–21458. Bibcode:2010PNAS..10721453B. doi: 10.1073/pnas.1014982107 . ISSN 0027-8424. PMC 3003128 . PMID 21115845.
- ↑ Barbatti, Mario; Borin, Antonio Carlos; Ullrich, Susanne (2014), Barbatti, Mario; Borin, Antonio Carlos; Ullrich, Susanne (eds.), "Photoinduced Processes in Nucleic Acids", Photoinduced Phenomena in Nucleic Acids I, vol. 355, Springer International Publishing, pp. 1–32, doi:10.1007/128_2014_569, ISBN 978-3-319-13370-6, PMID 25381199
- ↑ Boulanger, Eliot; Anoop, Anakuthil; Nachtigallova, Dana; Thiel, Walter; Barbatti, Mario (2013-07-29). "Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN". Angewandte Chemie International Edition. 52 (31): 8000–8003. doi:10.1002/anie.201303246. hdl:11858/00-001M-0000-0014-A349-D. PMID 23784979.
- ↑ Barbatti, Mario (2014-07-23). "Photorelaxation Induced by Water–Chromophore Electron Transfer". Journal of the American Chemical Society. 136 (29): 10246–10249. doi:10.1021/ja505387c. hdl: 11858/00-001M-0000-0024-A591-C . ISSN 0002-7863. PMID 25010652. S2CID 207112041.
- ↑ de Medeiros, Vanessa C.; de Andrade, Railton B.; Leitão, Ezequiel F. V.; Ventura, Elizete; Bauerfeldt, Glauco F.; Barbatti, Mario; do Monte, Silmar A. (2016-01-13). "Photochemistry of CH3Cl: Dissociation and CH···Cl Hydrogen Bond Formation" (PDF). Journal of the American Chemical Society. 138 (1): 272–280. doi:10.1021/jacs.5b10573. ISSN 0002-7863. PMID 26653216.
- ↑ Fazzi, Daniele; Barbatti, Mario; Thiel, Walter (2017-10-05). "Hot and Cold Charge-Transfer Mechanisms in Organic Photovoltaics: Insights into the Excited States of Donor/Acceptor Interfaces" (PDF). The Journal of Physical Chemistry Letters. 8 (19): 4727–4734. doi:10.1021/acs.jpclett.7b02144. hdl:11858/00-001M-0000-002E-9D60-C. ISSN 1948-7185. PMID 28903560. S2CID 46862226.
- ↑ Barbatti, Mario; Nascimento, Marco Antonio Chaer (2012-10-05). "Does the H+5 hydrogen cluster exist in dense interstellar clouds?". International Journal of Quantum Chemistry. 112 (19): 3169–3173. doi:10.1002/qua.24110.
- ↑ Pereira Rodrigues, Gessenildo; Lopes de Lima, Thayana Maria; de Andrade, Railton Barbosa; Ventura, Elizete; do Monte, Silmar Andrade; Barbatti, Mario (2019-03-14). "Photoinduced Formation of H-Bonded Ion Pair in HCFC-133a" (PDF). The Journal of Physical Chemistry A. 123 (10): 1953–1961. Bibcode:2019JPCA..123.1953P. doi:10.1021/acs.jpca.8b12482. ISSN 1089-5639. PMID 30786711. S2CID 73466468.
- ↑ Barbatti, Mario (2020-08-11). "Simulation of Excitation by Sunlight in Mixed Quantum-Classical Dynamics". Journal of Chemical Theory and Computation. 16 (8): 4849–4856. doi:10.1021/acs.jctc.0c00501. ISSN 1549-9618. PMC 7426902 . PMID 32579345.
- ↑ "Interview with Mario Barbatti, the first Brazilian ERC Advanced grant awardee". EURAXESS. 2019-05-30. Retrieved 2019-12-29.
- ↑ Dral, Pavlo O.; Barbatti, Mario; Thiel, Walter (2018-10-04). "Nonadiabatic Excited-State Dynamics with Machine Learning". The Journal of Physical Chemistry Letters. 9 (19): 5660–5663. doi:10.1021/acs.jpclett.8b02469. ISSN 1948-7185. PMC 6174422 . PMID 30200766.
- ↑ "Electrons on the fast lane". News-Medical.net. 2009-02-23. Retrieved 2019-12-29.
- ↑ "How do DNA components resist damaging UV exposure?". ScienceDaily. Retrieved 2019-12-29.
- ↑ "Physicists uncover new data on adenine, a crucial building block of life". ScienceDaily. Retrieved 2019-12-29.
- ↑ "Scientists Discover How Life is Formed Out of Dead Matter". Nature World News. 2013-06-25. Retrieved 2019-12-29.
- ↑ "Excited, but cold: Scientists unveil the secret of a reaction for prebiotic synthesis of organic matter". phys.org. Retrieved 2019-12-29.
- ↑ "Spark of life". The Telegraph. Retrieved 2019-12-29.
- ↑ Solvent Matters! Effect of Environment on Photorelaxation (2014-08-06). "Spotlights on Recent JACS Publications". Journal of the American Chemical Society. 136 (31): 10821–10822. doi:10.1021/ja5076508. ISSN 0002-7863.
- ↑ "Quand Chlore et Hydrogène vivent une liaison électrique". Archives des actualités de l'INC - CNRS (in French). Retrieved 2019-12-29.