Related Research Articles

Tetralogy of Fallot (TOF), formerly known as Steno-Fallot tetralogy, is a congenital heart defect characterized by four specific cardiac defects. Classically, the four defects are:

Hirschsprung's disease is a birth defect in which nerves are missing from parts of the intestine. The most prominent symptom is constipation. Other symptoms may include vomiting, abdominal pain, diarrhea and slow growth. Most children develop signs and symptoms shortly after birth. However, others may be diagnosed later in infancy or early childhood. About half of all children with Hirschsprung's disease are diagnosed in the first year of life. Complications may include enterocolitis, megacolon, bowel obstruction and intestinal perforation.

An omphalocele or omphalocoele, also known as an exomphalos, is a rare abdominal wall defect. Beginning at the 6th week of development, rapid elongation of the gut and increased liver size reduces intra abdominal space, which pushes intestinal loops out of the abdominal cavity. Around 10th week, the intestine returns to the abdominal cavity and the process is completed by the 12th week. Persistence of intestine or the presence of other abdominal viscera in the umbilical cord results in an omphalocele.
Miller–Dieker syndrome, Miller–Dieker lissencephaly syndrome (MDLS), and chromosome 17p13.3 deletion syndrome is a micro deletion syndrome characterized by congenital malformations. Congenital malformations are physical defects detectable in an infant at birth which can involve many different parts of the body including the brain, hearts, lungs, liver, bones, or intestinal tract. MDS is a contiguous gene syndrome – a disorder due to the deletion of multiple gene loci adjacent to one another. The disorder arises from the deletion of part of the small arm of chromosome 17p, leading to partial monosomy. There may be unbalanced translocations, or the presence of a ring chromosome 17.

Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence is often normal. Complications of NS can include leukemia.
An imperforate hymen is a congenital disorder where a hymen without an opening completely obstructs the vagina. It is caused by a failure of the hymen to perforate during fetal development. It is most often diagnosed in adolescent girls when menstrual blood accumulates in the vagina and sometimes also in the uterus. It is treated by surgical incision of the hymen.

Dandy–Walker malformation (DWM), also known as Dandy–Walker syndrome (DWS), is a rare congenital brain malformation in which the part joining the two hemispheres of the cerebellum does not fully form, and the fourth ventricle and space behind the cerebellum are enlarged with cerebrospinal fluid. Most of those affected develop hydrocephalus within the first year of life, which can present as increasing head size, vomiting, excessive sleepiness, irritability, downward deviation of the eyes and seizures. Other, less common symptoms are generally associated with comorbid genetic conditions and can include congenital heart defects, eye abnormalities, intellectual disability, congenital tumours, other brain defects such as agenesis of the corpus callosum, skeletal abnormalities, an occipital encephalocele or underdeveloped genitalia or kidneys. It is sometimes discovered in adolescents or adults due to mental health problems.

Simpson–Golabi–Behmel syndrome (SGBS) is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.
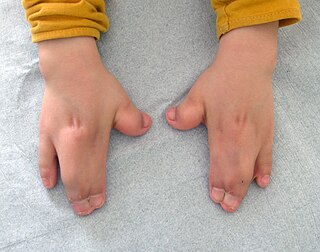
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.
Interrupted aortic arch is a very rare heart defect in which the aorta is not completely developed. There is a gap between the ascending and descending thoracic aorta. In a sense it is the complete form of a coarctation of the aorta. Almost all patients also have other cardiac anomalies, including a ventricular septal defect (VSD), aorto-pulmonary window, and truncus arteriosus. There are three types of interrupted aortic arch, with type B being the most common. Interrupted aortic arch is often associated with DiGeorge syndrome.
Hematocolpos is a medical condition in which the vagina is pooled with menstrual blood due to multiple factors leading to the blockage of menstrual blood flow. The medical definition of hematocolpos is "an accumulation of blood within the vagina". It is often caused by the combination of menstruation with an imperforate hymen. It is sometimes seen in Robinow syndrome, uterus didelphys, or other vaginal anomalies.

3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.

Hypoplastic right heart syndrome (HRHS) is a congenital heart defect in which the structures on the right side of the heart, particularly the right ventricle, are underdeveloped. This defect causes inadequate blood flow to the lungs, and thus a cyanotic infant.
L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.
Chest pain in children is the pain felt in the chest by infants, children and adolescents. In most cases the pain is not associated with the heart. It is primarily identified by the observance or report of pain by the infant, child or adolescent by reports of distress by parents or caregivers. Chest pain is not uncommon in children. Many children are seen in ambulatory clinics, emergency departments and hospitals and cardiology clinics. Most often there is a benign cause for the pain for most children. Some have conditions that are serious and possibly life-threatening. Chest pain in pediatric patients requires careful physical examination and a detailed history that would indicate the possibility of a serious cause. Studies of pediatric chest pain are sparse. It has been difficult to create evidence-based guidelines for evaluation.

Vaginal anomalies are abnormal structures that are formed during the prenatal development of the female reproductive system and are rare congenital defects that result in an abnormal or absent vagina.

Acyl-CoA oxidase deficiency is a rare disorder that leads to significant damage and deterioration of nervous system functions (neurodegeneration). It is caused by pathogenic variants in ACOX1, which codes for the production of an enzyme called peroxisomal straight-chain acyl-CoA oxidase (ACOX1). This specific enzyme is responsible for the breakdown of very long chain fatty acids (VLCFAs).
Caudal duplication, is a rare congenital disorder in which various structures of the caudal region, embryonic cloaca, and neural tube exhibit a spectrum of abnormalities such as duplication and malformations. The exact causes of the condition is unknown, though there are several theories implicating abnormal embryological development as a cause for the condition. Diagnosis is often made during prenatal development of the second trimester through anomaly scans or immediately after birth. However, rare cases of adulthood diagnosis has also been observed. Treatment is often required to correct such abnormalities according to the range of symptoms present, whilst treatment options vary from conservative expectant management to resection of caudal tissue to restore normal function or appearance. As a rare congenital disorder, the prevalence at birth is less than 1 per 100,000 with less than 100 cases reported worldwide.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.
References
- 1 2 3 Dosedla, Erik; Kacerovsky, Marian; Calda, Pavel (2011-03-01). "Prenatal diagnosis of hydrometrocolpos in a down syndrome fetus". Journal of Clinical Ultrasound. 39 (3): 169–171. doi:10.1002/jcu.20785. ISSN 1097-0096. PMID 21387330. S2CID 11211408.
- ↑ Yapar, E. G.; Ekici, E.; Aydogdu, T.; Senses, E.; Gökmen, O. (1996-12-18). "Diagnostic problems in a case with mucometrocolpos, polydactyly, congenital heart disease, and skeletal dysplasia". American Journal of Medical Genetics. 66 (3): 343–346. doi:10.1002/(SICI)1096-8628(19961218)66:3<343::AID-AJMG19>3.0.CO;2-M. ISSN 0148-7299. PMID 8985498.
- ↑ Babcock, Diane S. (January 1989). Neonatal and pediatric ultrasonography. Churchill Livingstone. ISBN 9780443086069.
- ↑ Saclarides, Theodore J.; Myers, Jonathan A.; Millikan, Keith W. (2015-01-02). Common Surgical Diseases: An Algorithmic Approach to Problem Solving. Springer. ISBN 9781493915651.
- ↑ Kaiser, Georges L. (2012-12-13). Symptoms and Signs in Pediatric Surgery. Springer Science & Business Media. ISBN 9783642311611.
- ↑ Stevenson, Roger E. (2015-10-27). Human Malformations and Related Anomalies. Oxford University Press. ISBN 9780199386031.
| | This women's health related article is a stub. You can help Wikipedia by expanding it. |