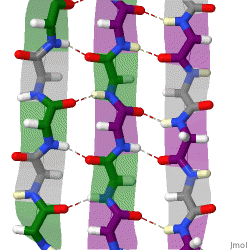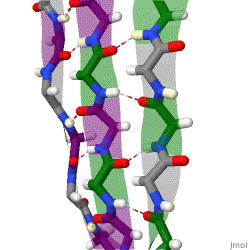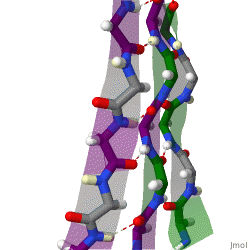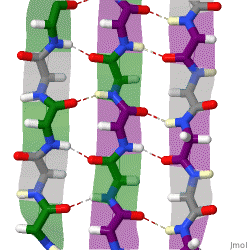
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.
DnaG is a bacterial DNA primase and is encoded by the dnaG gene. The enzyme DnaG, and any other DNA primase, synthesizes short strands of RNA known as oligonucleotides during DNA replication. These oligonucleotides are known as primers because they act as a starting point for DNA synthesis. DnaG catalyzes the synthesis of oligonucleotides that are 10 to 60 nucleotides long, however most of the oligonucleotides synthesized are 11 nucleotides. These RNA oligonucleotides serve as primers, or starting points, for DNA synthesis by bacterial DNA polymerase III. DnaG is important in bacterial DNA replication because DNA polymerase cannot initiate the synthesis of a DNA strand, but can only add nucleotides to a preexisting strand. DnaG synthesizes a single RNA primer at the origin of replication. This primer serves to prime leading strand DNA synthesis. For the other parental strand, the lagging strand, DnaG synthesizes an RNA primer every few kilobases (kb). These primers serve as substrates for the synthesis of Okazaki fragments.

The Rossmann fold is a tertiary fold found in proteins that bind nucleotides, such as enzyme cofactors FAD, NAD+, and NADP+. This fold is composed of alternating beta strands and alpha helical segments where the beta strands are hydrogen bonded to each other forming an extended beta sheet and the alpha helices surround both faces of the sheet to produce a three-layered sandwich. The classical Rossmann fold contains six beta strands whereas Rossmann-like folds, sometimes referred to as Rossmannoid folds, contain only five strands. The initial beta-alpha-beta (bab) fold is the most conserved segment of the Rossmann fold. The motif is named after Michael Rossmann who first noticed this structural motif in the enzyme lactate dehydrogenase in 1970 and who later observed that this was a frequently occurring motif in nucleotide binding proteins.
A supersecondary structure is a compact three-dimensional protein structure of several adjacent elements of a secondary structure that is smaller than a protein domain or a subunit. Supersecondary structures can act as nucleations in the process of protein folding.
A DNA-binding domain (DBD) is an independently folded protein domain that contains at least one structural motif that recognizes double- or single-stranded DNA. A DBD can recognize a specific DNA sequence or have a general affinity to DNA. Some DNA-binding domains may also include nucleic acids in their folded structure.

In protein structures, a beta barrel(β barrel) is a beta sheet composed of tandem repeats that twists and coils to form a closed toroidal structure in which the first strand is bonded to the last strand. Beta-strands in many beta-barrels are arranged in an antiparallel fashion. Beta barrel structures are named for resemblance to the barrels used to contain liquids. Most of them are water-soluble outer membrane proteins and frequently bind hydrophobic ligands in the barrel center, as in lipocalins. Others span cell membranes and are commonly found in porins. Porin-like barrel structures are encoded by as many as 2–3% of the genes in Gram-negative bacteria. It has been shown that more than 600 proteins with various function such as oxidase, dismutase, and amylase contain the beta barrel structure.

A DNA clamp, also known as a sliding clamp, is a protein complex that serves as a processivity-promoting factor in DNA replication. As a critical component of the DNA polymerase III holoenzyme, the clamp protein binds DNA polymerase and prevents this enzyme from dissociating from the template DNA strand. The clamp-polymerase protein–protein interactions are stronger and more specific than the direct interactions between the polymerase and the template DNA strand; because one of the rate-limiting steps in the DNA synthesis reaction is the association of the polymerase with the DNA template, the presence of the sliding clamp dramatically increases the number of nucleotides that the polymerase can add to the growing strand per association event. The presence of the DNA clamp can increase the rate of DNA synthesis up to 1,000-fold compared with a nonprocessive polymerase.
Pilin refers to a class of fibrous proteins that are found in pilus structures in bacteria. These structures can be used for the exchange of genetic material, or as a cell adhesion mechanism. Although not all bacteria have pili or fimbriae, bacterial pathogens often use their fimbriae to attach to host cells. In Gram-negative bacteria, where pili are more common, individual pilin molecules are linked by noncovalent protein-protein interactions, while Gram-positive bacteria often have polymerized LPXTG pilin.

RNA-dependent RNA polymerase (RdRp) or RNA replicase is an enzyme that catalyzes the replication of RNA from an RNA template. Specifically, it catalyzes synthesis of the RNA strand complementary to a given RNA template. This is in contrast to typical DNA-dependent RNA polymerases, which all organisms use to catalyze the transcription of RNA from a DNA template.

In molecular biology, LSm proteins are a family of RNA-binding proteins found in virtually every cellular organism. LSm is a contraction of 'like Sm', because the first identified members of the LSm protein family were the Sm proteins. LSm proteins are defined by a characteristic three-dimensional structure and their assembly into rings of six or seven individual LSm protein molecules, and play a large number of various roles in mRNA processing and regulation.

In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.

Replication protein A (RPA) is the major protein that binds to single-stranded DNA (ssDNA) in eukaryotic cells. In vitro, RPA shows a much higher affinity for ssDNA than RNA or double-stranded DNA. RPA is required in replication, recombination and repair processes such as nucleotide excision repair and homologous recombination. It also plays roles in responding to damaged DNA.

In molecular biology, a Tudor domain is a conserved protein structural domain originally identified in the Tudor protein encoded in Drosophila. The Tudor gene was found in a Drosophila screen for maternal factors that regulate embryonic development or fertility. Mutations here are lethal for offspring, inspiring the name Tudor, as a reference to the Tudor King Henry VIII and the several miscarriages experienced by his wives.

T7 DNA polymerase is an enzyme used during the DNA replication of the T7 bacteriophage. During this process, the DNA polymerase “reads” existing DNA strands and creates two new strands that match the existing ones. The T7 DNA polymerase requires a host factor, E. coli thioredoxin, in order to carry out its function. This helps stabilize the binding of the necessary protein to the primer-template to improve processivity by more than 100-fold, which is a feature unique to this enzyme. It is a member of the Family A DNA polymerases, which include E. coli DNA polymerase I and Taq DNA polymerase.
In molecular biology, proteins are generally thought to adopt unique structures determined by their amino acid sequences. However, proteins are not strictly static objects, but rather populate ensembles of conformations. Transitions between these states occur on a variety of length scales and time scales , and have been linked to functionally relevant phenomena such as allosteric signaling and enzyme catalysis.

In the fields of geometry and biochemistry, a triple helix is a set of three congruent geometrical helices with the same axis, differing by a translation along the axis. This means that each of the helices keeps the same distance from the central axis. As with a single helix, a triple helix may be characterized by its pitch, diameter, and handedness. Examples of triple helices include triplex DNA, triplex RNA, the collagen helix, and collagen-like proteins.

EF-P is an essential protein that in bacteria stimulates the formation of the first peptide bonds in protein synthesis. Studies show that EF-P prevents ribosomes from stalling during the synthesis of proteins containing consecutive prolines. EF-P binds to a site located between the binding site for the peptidyl tRNA and the exiting tRNA. It spans both ribosomal subunits with its amino-terminal domain positioned adjacent to the aminoacyl acceptor stem and its carboxyl-terminal domain positioned next to the anticodon stem-loop of the P site-bound initiator tRNA. The EF-P protein shape and size is very similar to a tRNA and interacts with the ribosome via the exit “E” site on the 30S subunit and the peptidyl-transferase center (PTC) of the 50S subunit. EF-P is a translation aspect of an unknown function, therefore It probably functions indirectly by altering the affinity of the ribosome for aminoacyl-tRNA, thus increasing their reactivity as acceptors for peptidyl transferase.

In molecular biology, protein fold classes are broad categories of protein tertiary structure topology. They describe groups of proteins that share similar amino acid and secondary structure proportions. Each class contains multiple, independent protein superfamilies.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.
