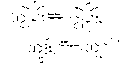Pancratistatin occurs naturally in Hawaiian spider lily, a flowering plant within the family Amaryllidaceae. Pancratistatin is mostly found in the bulb tissues of spider lilies. It has been shown that the enrichment of atmospheric CO2 can enhance the production of antiviral secondary metabolites, including pancratistatin, in these plants.[3] Pancratistatin can be isolated from the tropical bulbs of Hymenocallis littoralis in the order of 100 to 150mg/kg when bulbs are obtained from the wild type in Hawaii. However, the compound has to be commercially extracted from field- and greenhouse-grown bulbs or from tissue cultures cultivated, for example, in Arizona, which generate lower levels of pancratistatin (a maximum of 22mg/kg) even in the peak month of October. After October, when the bulb becomes dormant, levels of pancratistatin drop, down to only 4mg/kg by May. Field-grown bulbs, which show monthly changes in pancratistatin content, generate somewhat smaller amounts (2–5mg/kg) compared to those grown in greenhouses cultivated over the same period.[4] There are about 40 different spider lily species worldwide and they are mainly native to the Andes of South America.
Schoals Spider Lilly
Pharmaceutical research
Pancratistatin is thought to have potential as a basis for the development of new pharmaceuticals,[5] particularly in the field of cancer treatment.[6]
Biosynthesis
Comparison of pancratistatin and narciclasine
Although there may not be a precise elucidation of pancratistatin biological synthesis, there have been speculations on biosynthesis of narciclasine and lycoricidine that are very similar to pancratistatin in terms of structure. The biosynthesis is accomplished via synthesis from O-methylnorbelladine by para-para phenol coupling to obtain vittatine as an intermediate. Subsequent elimination of two carbon atoms and hydroxylations of vittatine then leads to narciclasine.[7]
Biosynthesis of the pancratistatin-like chemical compound narciclasine
Total synthesis
The first total synthesis [8] of racemic (+/-)-pancratistatin was reported by Samuel Danishefsky and Joung Yon Lee, which involved a very complex and long (40 steps) total synthesis. According to Danishefsky and Joung, there were several weak steps in this synthesis that gave rise to a disappointing low synthetic yield. Amongst the most challenging issues, the Moffatt transposition and the orthoamide problem, which required a blocking maneuver to regiospecifically distinguish the C, hydroxyl group for rearrangement were considered to be the severe cases. However, both Danishevsky and Yon Lee stated that their approach towards the PST total synthesis was not without merit and believed that their work would interest other medicinal scientists to construct a much more practical and efficient way for PST total synthesis.[9][10]
Danishevsky and Joung total synthesis of racemic pancratistatin
The work of Danishevsky and Joung provided the foundation for another total synthesis of PST, which was propounded by Li, M. in 2006. This method employed a more sophisticated approach, starting out with pinitol that has stereocenters which are exactly the same as the ones in the C-ring of pancratistatin.[11] Protection of the diol functions of compound 30 gave compound 31. The free hydroxyl of this was subsequently substituted by an azide to give 32. After removal of the silyl function, a cyclic sulfate was installed to obtain product 33. The Staudinger reaction gave the free amine 34 from azide 33. The coupling reaction between 34 and 35 gave compound 36 with a moderate yield. Methoxymethyl protection of both the amide and the free phenol gave compound 37. Treatment of this latter product with t-BuLi followed by addition of cerium chloride gave compound 38. Full deprotection of 38 by BBr3 and methanol afforded pancratistatin 3 in 12 steps from commercially available pinitol with an overall yield of 2.3% 20.[12]
The abstract of the Stereocontrolled synthesis of pancratistatin
Streocontrolled synthesis of pancratistatin
The most recent and shortest synthesis of pancratistatin was accomplished by David Sarlah and co workers, completing the asymmetric synthesis of (+)-pancratistatin and (+)-7-deoxypancratistatin in 7 and 6 steps respectively.[13] The key step of this synthesis was the Nickel catalyzed dearomatization of benzene which directly installed the amine and catachol ring in 98:2 er. Epoxidation then dihydroxylation of the resulting diene afforded the 4 hydroxyl groups. The synthesis was completed by deprotection of the amine and a Cobalt catalyzed CO insertion to furnish the lactam. (+)-7-deoxypancratistatin can then be directly oxidized in a 62% yield to give (+)-pancratistatin. This synthesis yielded multiple grams of the final product which may be essential in the biological evaluation of pancratistatin and analogues.
A very recent approach to a stereocontrolled pancratistatin synthesis was accomplished by Sanghee Kim from the National University of Seoul, in which Claisen rearrangement of dihydropyranethlyene and a cyclic sulfate elimination reaction were employed 21.
The B ring of the phenanthridone (three-membered nitrogen heterocyclic ring) is formed using the Bischler-Napieralski reaction. The n precursor 3 with its stereocenters in the C ring is stereoselectively synthesized from the cis-disubstituted cyclohexene 4. The presence of unsaturated carbonyl in compound 4 suggested the use of a Claisen rearrangement of 3,4-dihydro-2H-pyranylethylene.[14]
The synthesis starts with the treatment of 6 with excess trimethyl phosphate. This reaction provides phosphate 7 in 97% yield. Using the Horner-Wadsworth-Emmons reaction between 7 and acrolein dimmer 8 in the presence of LHMDS in THF forms (E)-olefin 5 with very high stereoselectivity in 60% yield. Only less than 1% of (Z)-olefin was detected in the final product. The Claisen rearrangement of dihydropyranethylene forms the cis-distributed cyclohexene as a single isomer in 78% yield.
The next step of the synthesis involves the oxidation of aldehyde of compound 4 using NaClO2 to the corresponding carboxylic acid 9 in 90% yield. Iodolactonization of 9 and subsequent treatment with DBU in refluxing benzene gives rise to the bicyclic lacytone in 78% yield. Methanolysis of lactone 10 with NaOMe forms a mixture of hydroxyl ester 11 and its C-4a epimer (pancratistatin numbering). Saponification of the methyl ester 11 with LiOH was followed by a Curtius rearrangement of the resulting acid 12 with diphenylphosphoryl azide in refluxing toluene to afford an isocyanate intermediate, treatment of which with NaOMe/MeOH forms the corresponding carbamate 13 in 82% yield.
The next steps of the synthesis involve the regioselective elimination of the C-3 hydroxyl group and subsequent unsaturation achieved by cyclic sulfate elimination. Diol 16 needs to be treated with thionyl chloride and further oxidation with RuCl3 provides the cyclic sulfate 17 in 83% yield.[15] Treatment of cyclic sulfate with DBU yields the desired allylic alcohol 18 (67% yield).
Reaction with OsO4 forms the single isomer 19 in 88% yield. Peracetylation of 19 (77% yield) accompanied by Banwell’s modified Bischler-Napieralski reaction forms the compound 20 with a small amount of isomer 21 ( 7:1 regioselectivity). The removal of protecting groups with NaOMe/MeOH forms pancratistatin in 83%.
↑Shnyder SD, Cooper PA, Millington NJ, Gill JH, Bibby MC (March 2008). "Sodium pancratistatin 3,4-o-cyclic phosphate, a water-soluble synthetic derivative of pancratistatin, is highly effective in a human colon tumor model". Journal of Natural Products. 71 (3): 321–324. doi:10.1021/np070477p. PMID18154271.
↑Ziska LH, Emche SD, Johnson EL, George KA, Reed DR, Sicher RC (October 2005). "Alterations in the production and concentration of selected alkaloids as a function of rising atmospheric carbon dioxide and air temperature: implications for ethno-pharmacology". Global Change Biology. 11 (10): 1798–1807. Bibcode:2005GCBio..11.1798Z. doi:10.1111/j.1365-2486.2005.001029.x. S2CID85828604.
↑Fuganti C, Staunton J, Battersby AR (1971). "The biosynthesis of narciclasine". Journal of the Chemical Society D: Chemical Communications. 19 (19): 1154–1155. doi:10.1039/C29710001154.
↑Danishefsky S, Lee JY (June 1989). "Total synthesis of (.+-.)-pancratistatin". Journal of the American Chemical Society. 111 (13): 4829–4837. doi:10.1021/ja00195a039.
↑Li M, Wu A, Zhou P (May 2006). "A concise synthesis of (+)-pancratistatin using pinitol as a chiral building block". Tetrahedron Letters. 47 (22): 3707–3710. doi:10.1016/j.tetlet.2006.03.138.
↑Kim S, Ko H, Kim E, Kim D (April 2002). "Stereocontrolled total synthesis of pancratistatin". Organic Letters. 4 (8): 1343–1345. doi:10.1021/ol0256419. PMID11950358.
↑Image gallery: a: TIPDSCl2, imidazole, DMAP, DMF, 24%. b: DMP, p-TsOH, acetone, 81%. c: PPh3, DEAD, CH3SO3H, CH2Cl2, 0°C to r.t. then NaN3, DMF, 60°C, 72%. d: TBAF, THF, 0°C to r.t., 100%. e: SOCl2, Et3N, CH2Cl2, 0°C. f: NaIO4, RuCl3, aq CH3CN, 87% (more than two steps). g: PPh3, aq THF, 0°C to r.t., 94%. h: Et2O, 35, 0°C, 64%. i: K2CO3, MOMCl, DMF, 84%. j: t-BuLi, CeCl3, ultrasound, THF, −78°C to r.t., 72%. k: BBr3, CH2Cl2, −78°C to 0°C, 1 hour then MeOH, −78°C to 0°C, 2 hours, 52%.
↑Shin KJ, Moon HR, George C, Marquez VE (April 2000). "Construction of the bicyclo[3.1.0]hexane template of a conformationally locked carbocyclic adenosine via an olefin keto-carbene cycloaddition". The Journal of Organic Chemistry. 65 (7): 2172–8. doi:10.1021/jo9917691. PMID10774042.
↑Winkler JD, Kim S, Harrison S, Lewin NE, Blumberg PM (January 1999). "Synthesis and Biological Evaluation of Highly Functionalized Analogues of Ingenol". Journal of the American Chemical Society. 121 (2): 296–300. doi:10.1021/ja9741842.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.