Related Research Articles

Yeast artificial chromosomes (YACs) are genetically engineered chromosomes derived from the DNA of the yeast, Saccharomyces cerevisiae, which is then ligated into a bacterial plasmid. By inserting large fragments of DNA, from 100–1000 kb, the inserted sequences can be cloned and physically mapped using a process called chromosome walking. This is the process that was initially used for the Human Genome Project, however due to stability issues, YACs were abandoned for the use of bacterial artificial chromosome
Comparative genomic hybridization (CGH) is a molecular cytogenetic method for analysing copy number variations (CNVs) relative to ploidy level in the DNA of a test sample compared to a reference sample, without the need for culturing cells. The aim of this technique is to quickly and efficiently compare two genomic DNA samples arising from two sources, which are most often closely related, because it is suspected that they contain differences in terms of either gains or losses of either whole chromosomes or subchromosomal regions. This technique was originally developed for the evaluation of the differences between the chromosomal complements of solid tumor and normal tissue, and has an improved resolution of 5–10 megabases compared to the more traditional cytogenetic analysis techniques of giemsa banding and fluorescence in situ hybridization (FISH) which are limited by the resolution of the microscope utilized.
Chromosome jumping is a tool of molecular biology that is used in the physical mapping of genomes. It is related to several other tools used for the same purpose, including chromosome walking.
Genetics, a discipline of biology, is the science of heredity and variation in living organisms.

Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only particular parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.
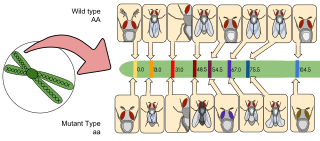
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
A genomic library is a collection of overlapping DNA fragments that together make up the total genomic DNA of a single organism. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. In order to construct a genomic library, the organism's DNA is extracted from cells and then digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector using DNA ligase. Next, the vector DNA can be taken up by a host organism - commonly a population of Escherichia coli or yeast - with each cell containing only one vector molecule. Using a host cell to carry the vector allows for easy amplification and retrieval of specific clones from the library for analysis.
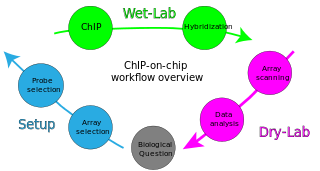
ChIP-on-chip is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest. As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.

Chromosome conformation capture techniques are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome. Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide. Interaction frequencies may be analyzed directly, or they may be converted to distances and used to reconstruct 3-D structures.
The following outline is provided as an overview of and topical guide to genetics:

Tiling arrays are a subtype of microarray chips. Like traditional microarrays, they function by hybridizing labeled DNA or RNA target molecules to probes fixed onto a solid surface.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Diversity Arrays Technology (DArT) is a high-throughput genetic marker technique that can detect allelic variations to provide comprehensive genome coverage without any DNA sequence information for genotyping and other genetic analysis. The general steps involve reducing the complexity of the genomic DNA with specific restriction enzymes, choosing diverse fragments to serve as representations for the parent genomes, amplify via polymerase chain reaction (PCR), inserting fragments into a vector to be placed as probes within a microarray, and then fluorescent targets from a reference sequence will be allowed to hybridize with probes and put through an imaging system. The objective is to identify and quantify various forms of DNA polymorphism within genomic DNA of sampled species.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.

Chromatin immunoprecipitation (ChIP) is a type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites, and possibly define cistromes. ChIP also aims to determine the specific location in the genome that various histone modifications are associated with, indicating the target of the histone modifiers. ChIP is crucial for the advancements in the field of epigenomics and learning more about epigenetic phenomena.
Radiation hybrid mapping is a technique for mapping mammalian chromosomes.

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow the analysis of large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two "ends" is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.

Single cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.

Vectorette PCR is a variation of polymerase chain reaction (PCR) designed in 1988. The original PCR was created and also patented during the 1980s. Vectorette PCR was first noted and described in an article in 1990 by John H. Riley and his team. Since then, multiple variants of PCR have been created. Vectorette PCR focuses on amplifying a specific sequence obtained from an internal sequence that is originally known until the fragment end. Multiple researches have taken this method as an opportunity to conduct experiments in order to uncover the potential uses that can be derived from Vectorette PCR.

Hi-C is a high-throughput genomic and epigenomic technique to capture chromatin conformation (3C). In general, Hi-C is considered as a derivative of a series of chromosome conformation capture technologies, including but not limited to 3C, 4C, and 5C. Hi-C comprehensively detects genome-wide chromatin interactions in the cell nucleus by combining 3C and next-generation sequencing (NGS) approaches and has been considered as a qualitative leap in C-technology development and the beginning of 3D genomics.
References
- ↑ Brown TA (2002). "Mapping Genomes". Genomes. Oxford: Wiley-Liss – via NCBI.
- ↑ Alizadeh, F.; Karp, R. M.; Weisser, D. K.; Zweig, G. (1995). "Physical Mapping of Chromosomes Using Unique Probes". Journal of Computational Biology. 2 (2): 159–184. doi:10.1089/cmb.1995.2.159. PMID 7497125. S2CID 13628080.
- ↑ Griffiths AJ, Miller JH, Suzuki DT, et al. (2000). "Mapping human genes by using human–rodent somatic cell hybrids".
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 3 4 5 6 Strachan, T.; Read, Andrew P. (1999). Human molecular genetics 2 (2nd ed.). Singapore: J. Wiley. ISBN 978-0471330615. OCLC 154500424.
- 1 2 3 4 5 6 Reilly, Cavan. (2009). Statistics in human genetics and molecular biology. Boca Raton: Taylor & Francis. ISBN 9781420072631. OCLC 318585618.
- 1 2 Liu, Ben-Hui (1998). Statistical genomics: linkage, mapping, and QTL analysis. Boca Raton: CRC Press. ISBN 978-0849331664. OCLC 36423381.
- ↑ Glen A. Evans. "Physical mapping of complex genomes".
- 1 2 3 Genomics : the science and technology behind the Human Genome Project. Wiley. pp. 234–284. ISBN 978-0-471-22056-5.
- ↑ FARID ALIZADEH; RICHARD M. KARP; DEBORAH K. WEISSER; GEOFFREY ZWEIG (1995). "Physical Mapping of Chromosomes Using Unique Probes". Journal of Computational Biology. 2 (2): 159–84. doi:10.1089/cmb.1995.2.159. PMID 7497125. S2CID 13628080.
- 1 2 Böckenhauer, Hans-Joachim; Bongartz, Dirk (2007). Algorithmic Aspects of Bioinformatics. Springer Berlin Heidelberg. pp. 123–169. ISBN 9783540719120.
- ↑ MICHAEL FONSTEIN AND ROBERT HASELKORN (June 1995). "Physical Mapping of Bacterial Genomes". Journal of Bacteriology. 177 (12): 3361–3369. doi:10.1128/jb.177.12.3361-3369.1995. PMC 177037 . PMID 7768844.