Collection of the total genomic DNA from a single organism
A genomic library is a collection of overlapping DNA fragments that together make up the total genomic DNA of a single organism. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. In order to construct a genomic library, the organism's DNA is extracted from cells and then digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector using DNA ligase.[1] Next, the vector DNA can be taken up by a host organism - commonly a population of Escherichia coli or yeast - with each cell containing only one vector molecule. Using a host cell to carry the vector allows for easy amplification and retrieval of specific clones from the library for analysis.[2]
There are several kinds of vectors available with various insert capacities. Generally, libraries made from organisms with larger genomes require vectors featuring larger inserts, thereby fewer vector molecules are needed to make the library. Researchers can choose a vector also considering the ideal insert size to find the desired number of clones necessary for full genome coverage.[3]
Genomic libraries are commonly used for sequencing applications. They have played an important role in the whole genome sequencing of several organisms, including the human genome and several model organisms.[4][5]
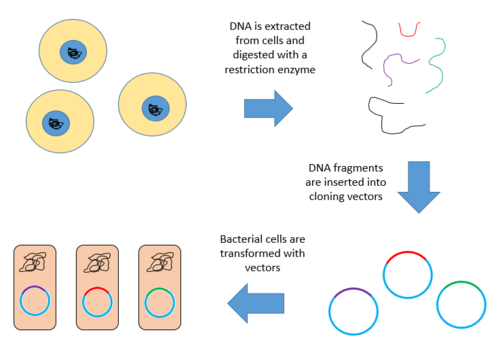
Construction of a genomic library involves creating many recombinant DNA molecules. An organism's genomic DNA is extracted and then digested with a restriction enzyme. For organisms with very small genomes (~10 kb), the digested fragments can be separated by gel electrophoresis. The separated fragments can then be excised and cloned into the vector separately. However, when a large genome is digested with a restriction enzyme, there are far too many fragments to excise individually. The entire set of fragments must be cloned together with the vector, and separation of clones can occur after. In either case, the fragments are ligated into a vector that has been digested with the same restriction enzyme. The vector containing the inserted fragments of genomic DNA can then be introduced into a host organism.[1]
Below are the steps for creating a genomic library from a large genome.
Digest the DNA with a restriction enzyme. This creates fragments that are similar in size, each containing one or more genes.
Insert the fragments of DNA into vectors that were cut with the same restriction enzyme. Use the enzyme DNA ligase to seal the DNA fragments into the vector. This creates a large pool of recombinant molecules.
These recombinant molecules are taken up by a host bacterium by transformation, creating a DNA library.[9][10]
Below is a diagram of the above outlined steps.
Genomic Library Construction
Determining titer of library
After a genomic library is constructed with a viral vector, such as lambda phage, the titer of the library can be determined. Calculating the titer allows researchers to approximate how many infectious viral particles were successfully created in the library. To do this, dilutions of the library are used to transformcultures of E. coli of known concentrations. The cultures are then plated on agar plates and incubated overnight. The number of viral plaques are counted and can be used to calculate the total number of infectious viral particles in the library. Most viral vectors also carry a marker that allows clones containing an insert to be distinguished from those that do not have an insert. This allows researchers to also determine the percentage of infectious viral particles actually carrying a fragment of the library.[11]
A similar method can be used to titer genomic libraries made with non-viral vectors, such as plasmids and BACs. A test ligation of the library can be used to transform E. coli. The transformation is then spread on agar plates and incubated overnight. The titer of the transformation is determined by counting the number of colonies present on the plates. These vectors generally have a selectable marker allowing the differentiation of clones containing an insert from those that do not. By doing this test, researchers can also determine the efficiency of the ligation and make adjustments as needed to ensure they get the desired number of clones for the library.[12]
Screening library
Colony Blot Hybridization
In order to isolate clones that contain regions of interest from a library, the library must first be screened. One method of screening is hybridization. Each transformed host cell of a library will contain only one vector with one insert of DNA. The whole library can be plated onto a filter over media. The filter and colonies are prepared for hybridization and then labeled with a probe.[13] The target DNA- insert of interest- can be identified by detection such as autoradiography because of the hybridization with the probe as seen below.
Another method of screening is with polymerase chain reaction (PCR). Some libraries are stored as pools of clones and screening by PCR is an efficient way to identify pools containing specific clones.[2]
Types of vectors
Genome size varies among different organisms and the cloning vector must be selected accordingly. For a large genome, a vector with a large capacity should be chosen so that a relatively small number of clones are sufficient for coverage of the entire genome. However, it is often more difficult to characterize an insert contained in a higher capacity vector.[3]
Below is a table of several kinds of vectors commonly used for genomic libraries and the insert size that each generally holds.
A plasmid is a double stranded circular DNA molecule commonly used for molecular cloning. Plasmids are generally 2 to 4 kilobase-pairs (kb) in length and are capable of carrying inserts up to 15kb. Plasmids contain an origin of replication allowing them to replicate inside a bacterium independently of the host chromosome. Plasmids commonly carry a gene for antibiotic resistance that allows for the selection of bacterial cells containing the plasmid. Many plasmids also carry a reporter gene that allows researchers to distinguish clones containing an insert from those that do not.[3]
Phage lambda (λ)
Phage λ is a double-stranded DNA virus that infects E. coli. The λ chromosome is 48.5kb long and can carry inserts up to 25kb. These inserts replace non-essential viral sequences in the λ chromosome, while the genes required for formation of viral particles and infection remain intact. The insert DNA is replicated with the viral DNA; thus, together they are packaged into viral particles. These particles are very efficient at infection and multiplication leading to a higher production of the recombinant λ chromosomes.[3] However, due to the smaller insert size, libraries made with λ phage may require many clones for full genome coverage.[14]
Cosmids
Cosmid vectors are plasmids that contain a small region of bacteriophage λ DNA called the cos sequence. This sequence allows the cosmid to be packaged into bacteriophage λ particles. These particles- containing a linearized cosmid- are introduced into the host cell by transduction. Once inside the host, the cosmids circularize with the aid of the host's DNA ligase and then function as plasmids. Cosmids are capable of carrying inserts up to 40kb in size.[2]
Bacteriophage P1 vectors
Bacteriophage P1 vectors can hold inserts 70 – 100kb in size. They begin as linear DNA molecules packaged into bacteriophage P1 particles. These particles are injected into an E. coli strain expressing Cre recombinase. The linear P1 vector becomes circularized by recombination between two loxP sites in the vector. P1 vectors generally contain a gene for antibiotic resistance and a positive selection marker to distinguish clones containing an insert from those that do not. P1 vectors also contain a P1 plasmid replicon, which ensures only one copy of the vector is present in a cell. However, there is a second P1 replicon- called the P1 lytic replicon- that is controlled by an inducible promoter. This promoter allows the amplification of more than one copy of the vector per cell prior to DNA extraction.[2]
bac vector
P1 artificial chromosomes
P1 artificial chromosomes (PACs) have features of both P1 vectors and Bacterial Artificial Chromosomes (BACs). Similar to P1 vectors, they contain a plasmid and a lytic replicon as described above. Unlike P1 vectors, they do not need to be packaged into bacteriophage particles for transduction. Instead they are introduced into E. coli as circular DNA molecules through electroporation just as BACs are.[2] Also similar to BACs, these are relatively harder to prepare due to a single origin of replication.[14]
Bacterial artificial chromosomes
Bacterial artificial chromosomes (BACs) are circular DNA molecules, usually about 7kb in length, that are capable of holding inserts up to 300kb in size. BAC vectors contain a replicon derived from E. coli F factor, which ensures they are maintained at one copy per cell.[4] Once an insert is ligated into a BAC, the BAC is introduced into recombination deficient strains of E. coli by electroporation. Most BAC vectors contain a gene for antibiotic resistance and also a positive selection marker.[2] The figure to the right depicts a BAC vector being cut with a restriction enzyme, followed by the insertion of foreign DNA that is re-annealed by a ligase. Overall, this is a very stable vector, but they may be hard to prepare due to a single origin of replication just like PACs.[14]
Yeast artificial chromosomes
Yeast artificial chromosomes (YACs) are linear DNA molecules containing the necessary features of an authentic yeast chromosome, including telomeres, a centromere, and an origin of replication. Large inserts of DNA can be ligated into the middle of the YAC so that there is an "arm" of the YAC on either side of the insert. The recombinant YAC is introduced into yeast by transformation; selectable markers present in the YAC allow for the identification of successful transformants. YACs can hold inserts up to 2000kb, but most YAC libraries contain inserts 250-400kb in size. Theoretically there is no upper limit on the size of insert a YAC can hold. It is the quality in the preparation of DNA used for inserts that determines the size limit.[2] The most challenging aspect of using YAC is the fact they are prone to rearrangement.[14]
How to select a vector
Vector selection requires one to ensure the library made is representative of the entire genome. Any insert of the genome derived from a restriction enzyme should have an equal chance of being in the library compared to any other insert. Furthermore, recombinant molecules should contain large enough inserts ensuring the library size is able to be handled conveniently.[14] This is particularly determined by the number of clones needed to have in a library. The number of clones to get a sampling of all the genes is determined by the size of the organism's genome as well as the average insert size. This is represented by the formula (also known as the Carbon and Clarke formula):[15]
is the desired probability that any fragment in the genome will occur at least once in the library created
is the fractional proportion of the genome in a single recombinant
can be further shown to be:
where,
is the insert size
is the genome size
Thus, increasing the insert size (by choice of vector) would allow for fewer clones needed to represent a genome. The proportion of the insert size versus the genome size represents the proportion of the respective genome in a single clone.[14] Here is the equation with all parts considered:
Vector selection example
The above formula can be used to determine the 99% confidence level that all sequences in a genome are represented by using a vector with an insert size of twenty thousand basepairs (such as the phage lambda vector). The genome size of the organism is three billion basepairs in this example.
clones
Thus, approximately 688,060 clones are required to ensure a 99% probability that a given DNA sequence from this three billion basepair genome will be present in a library using a vector with an insert size of twenty thousand basepairs.
Applications
After a library is created, the genome of an organism can be sequenced to elucidate how genes affect an organism or to compare similar organisms at the genome-level. The aforementioned genome-wide association studies can identify candidate genes stemming from many functional traits. Genes can be isolated through genomic libraries and used on human cell lines or animal models to further research.[17] Furthermore, creating high-fidelity clones with accurate genome representation and no stability issues would contribute well as intermediates for shotgun sequencing or the study of complete genes in functional analysis.[10]
Hierarchical sequencing
Whole genome shotgun sequencing versus Hierarchical shotgun sequencing
One major use of genomic libraries is hierarchichal shotgun sequencing, which is also called top-down, map-based or clone-by-clone sequencing. This strategy was developed in the 1980s for sequencing whole genomes before high throughput techniques for sequencing were available. Individual clones from genomic libraries can be sheared into smaller fragments, usually 500bp to 1000bp, which are more manageable for sequencing.[4] Once a clone from a genomic library is sequenced, the sequence can be used to screen the library for other clones containing inserts which overlap with the sequenced clone. Any new overlapping clones can then be sequenced forming a contig. This technique, called chromosome walking, can be exploited to sequence entire chromosomes.[2]
Whole genome shotgun sequencing is another method of genome sequencing that does not require a library of high-capacity vectors. Rather, it uses computer algorithms to assemble short sequence reads to cover the entire genome. Genomic libraries are often used in combination with whole genome shotgun sequencing for this reason. A high resolution map can be created by sequencing both ends of inserts from several clones in a genomic library. This map provides sequences of known distances apart, which can be used to help with the assembly of sequence reads acquired through shotgun sequencing.[4] The human genome sequence, which was declared complete in 2003, was assembled using both a BAC library and shotgun sequencing.[18][19]
Genome-wide association studies
Genome-wide association studies are general applications to find specific gene targets and polymorphisms within the human race. In fact, the International HapMap project was created through a partnership of scientists and agencies from several countries to catalog and utilize this data.[20] The goal of this project is to compare genetic sequences of different individuals to elucidate similarities and differences within chromosomal regions.[20] Scientists from all of the participating nations are cataloging these attributes with data from populations of African, Asian, and European ancestry. Such genome-wide assessments may lead to further diagnostic and drug therapies while also helping future teams focus on orchestrating therapeutics with genetic features in mind. These concepts are already being exploited in genetic engineering.[20] For example, a research team has actually constructed a PAC shuttle vector that creates a library representing two-fold coverage of the human genome.[17] This could serve as an incredible resource to identify genes, or sets of genes, causing disease. Moreover, these studies can serve as a powerful way to investigate transcriptional regulation as it has been seen in the study of baculoviruses.[21] Overall, advances in genome library construction and DNA sequencing has allowed for efficient discovery of different molecular targets.[5] Assimilation of these features through such efficient methods can hasten the employment of novel drug candidates.
References
12Losick, Richard; Watson, James D.; Tania A. Baker; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene. San Francisco: Pearson/Benjamin Cummings. ISBN978-0-8053-9592-1.
12345678Russell, David W.; Sambrook, Joseph (2001). Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory. ISBN978-0-87969-577-4.
12Osoegawa K, de Jong PJ, Frengen E, Ioannou PA (May 2001). "Construction of bacterial artificial chromosome (BAC/PAC) libraries". Curr Protoc Hum Genet. Chapter 5: 5.15.1–5.15.33. doi:10.1002/0471142905.hg0515s21. PMID18428289. S2CID8208834.
↑John R. McCarrey; Williams, Steven J.; Barton E. Slatko (2006). Laboratory investigations in molecular biology. Boston: Jones and Bartlett Publishers. ISBN978-0-7637-3329-2.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.