
Turner syndrome (TS), also known as 45,X, or 45,X0, is a genetic condition in which a female is partially or completely missing an X chromosome. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, those affected do not develop menstrual periods, or breasts without hormone treatment and are unable to have children without reproductive technology. Heart defects, diabetes, and low thyroid hormone occur in the disorder more frequently than average. Most people with Turner syndrome have normal intelligence; however, many have problems with spatial visualization that may be needed in order to learn mathematics. Vision and hearing problems also occur more often than average.

Septo-optic dysplasia (SOD), known also as de Morsier syndrome, is a rare congenital malformation syndrome that features a combination of the underdevelopment of the optic nerve, pituitary gland dysfunction, and absence of the septum pellucidum . Two or more of these features need to be present for a clinical diagnosis — only 30% of patients have all three. French-Swiss doctor Georges de Morsier first recognized the relation of a rudimentary or absent septum pellucidum with hypoplasia of the optic nerves and chiasm in 1956.
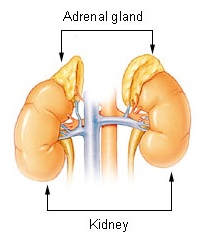
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal gland normally secretes glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. An adrenal crisis may occur if the body is subjected to stress, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.

Growth hormone deficiency (GHD), or human growth hormone deficiency, is a medical condition resulting from not enough growth hormone (GH). Generally the most noticeable symptom is that an individual attains a short height. Newborns may also present low blood sugar or a small penis size. In adults there may be decreased muscle mass, high cholesterol levels, or poor bone density.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.

Hypopituitarism is the decreased (hypo) secretion of one or more of the eight hormones normally produced by the pituitary gland at the base of the brain. If there is decreased secretion of one specific pituitary hormone, the condition is known as selective hypopituitarism. If there is decreased secretion of most or all pituitary hormones, the term panhypopituitarism is used.

Sheehan's syndrome, also known as postpartum pituitary gland necrosis, occurs when the pituitary gland is damaged due to significant blood loss and hypovolemic shock usually during or after childbirth leading to decreased functioning of the pituitary gland (hypopituitarism). The pituitary gland is an endocrine organ, meaning it produces certain hormones and is involved in the regulation of various other hormones. This gland is located in the brain and sits in a pocket of the sphenoid bone known as the sella turcica. The pituitary gland works in conjunction with the hypothalamus, and other endocrine organs to modulate numerous bodily functions including growth, metabolism, menstruation, lactation, and even the "fight-or-flight" response. These endocrine organs release hormones in very specific pathways, known as hormonal axes. For example, the release of a hormone in the hypothalamus will target the pituitary to trigger the release of a subsequent hormone, and the pituitary's released hormone will target the next organ in the pathway. Hence, damage to the pituitary gland can have downstream effects on any of the aforementioned bodily functions.
Optic nerve hypoplasia (ONH) is a medical condition arising from the underdevelopment of the optic nerve(s). This condition is the most common congenital optic nerve anomaly. The optic disc appears abnormally small, because not all the optic nerve axons have developed properly. It is often associated with endocrinopathies, developmental delay, and brain malformations. The optic nerve, which is responsible for transmitting visual signals from the retina to the brain, has approximately 1.2 million nerve fibers in the average person. In those diagnosed with ONH, however, there are noticeably fewer nerves.

Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms and the estimated prevalence rate in the general population is approximately 17%.
Kallmann syndrome (KS) is a genetic disorder that prevents a person from starting or fully completing puberty. Kallmann syndrome is a form of a group of conditions termed hypogonadotropic hypogonadism. To distinguish it from other forms of hypogonadotropic hypogonadism, Kallmann syndrome has the additional symptom of a total lack of sense of smell (anosmia) or a reduced sense of smell. If left untreated, people will have poorly defined secondary sexual characteristics, show signs of hypogonadism, almost invariably are infertile and are at increased risk of developing osteoporosis. A range of other physical symptoms affecting the face, hands and skeletal system can also occur.

Congenital hyperinsulinism is a medical term referring to a variety of congenital disorders in which hypoglycemia is caused by excessive insulin secretion. Congenital forms of hyperinsulinemic hypoglycemia can be transient or persistent, mild or severe. These conditions are present at birth and most become apparent in early infancy. Mild cases can be treated by frequent feedings, more severe cases can be controlled by medications that reduce insulin secretion or effects.

Pickardt syndrome denotes a rare form of tertiary hypothyroidism that is caused by interruption of the portal veins connecting hypothalamus and pituitary.It was characterized in 1972 and 1973 by Renate Pickardt and Rudolf Fahlbusch.

Empty sella syndrome is the condition when the pituitary gland shrinks or becomes flattened, filling the sella turcica with cerebrospinal fluid instead of the normal pituitary. It can be discovered as part of the diagnostic workup of pituitary disorders, or as an incidental finding when imaging the brain.
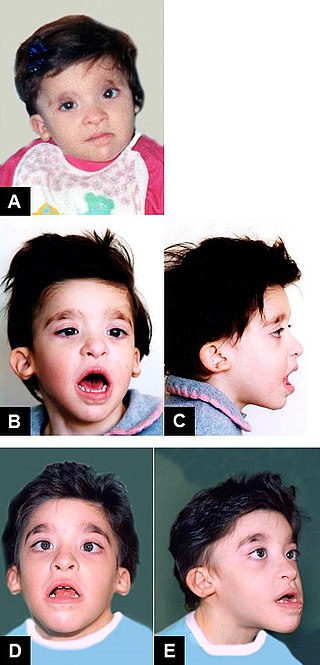
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998.

Dubowitz syndrome is a rare genetic disorder characterized by microcephaly, stunted growth, and a receding chin. Symptoms vary among patients, but other characteristics include a soft, high-pitched voice, partial webbing of the fingers and toes, palate deformations, genital abnormalities, language difficulties, and an aversion to crowds. The pathogenesis of the disease is yet to be identified, and no medical tests can definitively diagnose the disease. The primary method of diagnosis is to identify facial phenotypes. Since it was first described in 1965 by English physician Victor Dubowitz, over 140 cases have been reported worldwide. Although the majority of cases have been reported from the United States, Germany, and Russia, the disorder appears to affect both genders and all ethnicities equally.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.
Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.
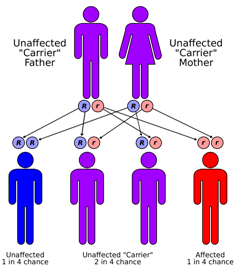
Sanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia, but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere. The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
