
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.

Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML), but these components do not always occur. DKC is characterized by short telomeres. The disease initially can affect the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.

Medical genetics is the branch of medicine that involves the diagnosis and management of hereditary disorders. Medical genetics differs from human genetics in that human genetics is a field of scientific research that may or may not apply to medicine, while medical genetics refers to the application of genetics to medical care. For example, research on the causes and inheritance of genetic disorders would be considered within both human genetics and medical genetics, while the diagnosis, management, and counselling people with genetic disorders would be considered part of medical genetics.

Blue diaper syndrome is a rare, autosomal recessive or X linked recessive metabolic disorder characterized in infants by bluish urine-stained diapers. It is also known as Drummond's syndrome, and hypercalcemia.
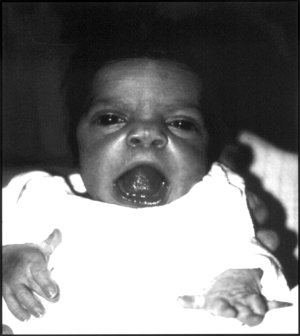
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.

Ectrodactyly–ectodermal dysplasia–cleft syndrome, or EEC, and also referred to as EEC syndrome and split hand–split foot–ectodermal dysplasia–cleft syndrome is a rare form of ectodermal dysplasia, an autosomal dominant disorder inherited as a genetic trait. EEC is characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefts. Other features noted in association with EEC include vesicoureteral reflux, recurrent urinary tract infections, obstruction of the nasolacrimal duct, decreased pigmentation of the hair and skin, missing or abnormal teeth, enamel hypoplasia, absent punctae in the lower eyelids, photophobia, occasional cognitive impairment and kidney anomalies, and conductive hearing loss.
Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.
Metaphyseal dysplasia, or Pyle disease, is a disorder of the bones. It is a rare disease in which the outer part of the shafts of long bones is thinner than normal and there is an increased chance of fractures. Its hallmark feature is an abnormality of the long bones in the arms and legs in which the ends (metaphyses) of the bones are abnormally broad; the shape of the bones resembles a boat oar or paddle. The broad metaphyses are due to enlargement of the spongy inner layer of bone. Although trabecular bone is expanded, the dense outermost layer of bone is thinner than normal. As a result, the bones are fragile and fracture easily. The bone abnormalities in the legs commonly cause knock knees in affected individuals.
Dermatopathia pigmentosa reticularis(DPR) is a rare, autosomal dominant congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. DPR is a non life-threatening disease that largely affects the skin, hair, and nails. It has also been identified as a keratin disorder. Historically, as of 1992, only 10 cases had been described in world literature; however, due to recent advances in genetic analysis, five additional families studied in 2006 have been added to the short list of confirmed cases.

Uncombable hair syndrome (UHS) is a rare structural anomaly of the hair with a variable degree of effect. It is characterized by hair that is silvery, dry, frizzy, wiry, and impossible to comb. It was first reported in the early 20th century. It typically becomes apparent between the ages of 3 months and 12 years. UHS has several names, including pili trianguli et canaliculi (Latin), cheveux incoiffables (French), and "spun-glass hair". This disorder is believed to be autosomal recessive in most instances, but there are a few documented cases where multiple family members display the trait in an autosomal dominant fashion. Based on the current scientific studies related to the disorder, the three genes that have been causally linked to UHS are PADI3, TGM3, and TCHH. These genes encode proteins important for hair shaft formation. Clinical symptoms of the disorder arise between 3 months and 12 years of age. The quantity of hair on the head does not change, but hair starts to grow more slowly and becomes increasingly "uncombable". To be clinically apparent, 50% of all scalp hair shafts must be affected by UHS. This syndrome only affects the hair shaft of the scalp and does not influence hair growth in terms of quantity, textural feel, or appearance on the rest of the body.
MORM syndrome is an autosomal recessive congenital disorder characterized by mental retardation, truncal obesity, retinal dystrophy, and micropenis". The disorder shares similar characteristics with Bardet–Biedl syndrome and Cohen syndrome, both of which are autosomal recessive genetic disorders. MORM syndrome can be distinguished from the above disorders because symptoms appear at a young age. The disorder is not dependent on sex of the offspring, both male and female offspring are equally likely to inherit the disorder.

Trichothiodystrophy (TTD) is an autosomal recessive inherited disorder characterised by brittle hair and intellectual impairment. The word breaks down into tricho – "hair", thio – "sulphur", and dystrophy – "wasting away" or literally "bad nourishment". TTD is associated with a range of symptoms connected with organs of the ectoderm and neuroectoderm. TTD may be subclassified into four syndromes: Approximately half of all patients with trichothiodystrophy have photosensitivity, which divides the classification into syndromes with or without photosensitivity; BIDS and PBIDS, and IBIDS and PIBIDS. Modern covering usage is TTD-P (photosensitive), and TTD.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
GRACILE syndrome is a very rare lethal autosomal recessive genetic disorder, one of the Finnish heritage diseases. GRACILE syndrome has also been found in the UK and Sweden, but not nearly as much as in Finland. It is caused by a mutation in the BCS1L gene and it occurs in approximately 1 out of 50,000 live births in Finnish people. To date, there have only been 32 cases of GRACILE syndrome reported.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
Eiken syndrome, also known as "Eiken skeletal dysplasia", is a rare autosomal bone dysplasia with a skeletal phenotype which has been described in a unique consanguineous family, where it segregates as a recessive trait. First described in 1985, the syndrome primarily affects the development of bones, leading to short stature, long limbs, and joint dislocations. Eiken syndrome is caused by mutations in the PTH1R gene, located on chromosome 3, and is involved in skeletal development.
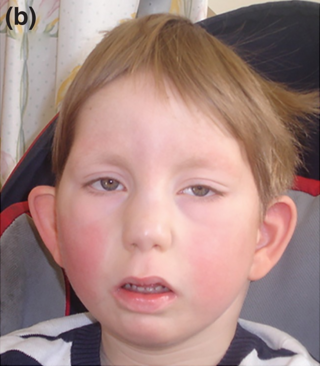
Okamoto syndrome (OS), also known as Au–Kline syndrome (AKS), is a very rare autosomal dominant genetic condition characterised by congenital hydronephrosis, low muscle tone, heart defects, intellectual disability and characteristic facial features. Those affected often have neurological and skeletal abnormalities, as well as frequent urinary tract infections. Language and walking are usually delayed. Facial features include prominent, downturned ears, an open, downturned mouth and drooping eyelids (ptosis).
Odontoonychodermal dysplasia is a rare genetic disorder which is characterized by systemic abnormalities of the teeth, the nails of the fingers and toes, the skin, the hair cells, and the sweat glands. It is a type of syndromic ectodermal dysplasia.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
