Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.

Ellis–Van Creveld syndrome is a rare genetic disorder of the skeletal dysplasia type.

Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B. These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues, and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death. The rare autosomal recessive neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.
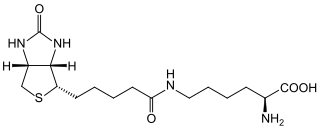
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.

2-hydroxyglutaric aciduria is a rare neurometabolic disorder characterized by the significantly elevated levels of hydroxyglutaric acid in one's urine. It is either autosomal recessive or autosomal dominant.
The GM1 gangliosidoses, usually shortened to GM1, are gangliosidoses caused by mutation in the GLB1 gene resulting in a deficiency of beta-galactosidase. The deficiency causes abnormal storage of acidic lipid materials in cells of the central and peripheral nervous systems, but particularly in the nerve cells, resulting in progressive neurodegeneration. GM1 is a rare lysosomal storage disorder with a prevalence of 1 to every 100,000 to 200,000 live births worldwide, although rates are higher in some regions.
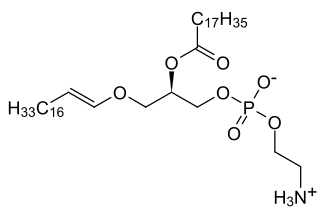
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by abnormally short arms and legs (rhizomelia), seizures, recurrent respiratory tract infections and congenital cataracts.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

Intermembrane lipid transfer protein VPS13B, also known as vacuolar protein sorting-associated 13B, and Cohen syndrome protein 1 is a protein that in humans is encoded by the VPS13B gene. It is a giant protein associated with the Golgi apparatus that is believed to be involved in post-Golgi apparatus sorting and trafficking. Mutations in the human VPS13B gene cause Cohen syndrome.

Lactosylceramide alpha-2,3-sialyltransferase is an enzyme that in humans is encoded by the ST3GAL5 gene.
De Barsy syndrome is a rare autosomal recessive genetic disorder. Symptoms include cutis laxa as well as other eye, musculoskeletal, and neurological abnormalities. It is usually progressive, manifesting side effects that can include clouded corneas, cataracts, short stature, dystonia, or progeria.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.

CDK13-related disorder, also known as congenital heart defects, dysmorphic facial features and intellectual developmental disorder (CHDFIDD), is a very rare autosomal dominant genetic condition characterised by congenital heart defects, intellectual disability and characteristic facial features. Those affected typically have motor and language delays, low muscle tone and gastrointestinal dysmotility. Facial features include a wide nasal bridge, widely-spaced eyes, prominent, low-set ears, a flat nose tip and a small mouth. Less common features include congenital spinal abnormalities, hearing loss or seizures.
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.

Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
SLC13A5 citrate transporter disorder, or SLC13A5 Epilepsy, is a rare genetic spectrum disorder that presents with neurological symptoms. Symptoms include severe seizures, ataxia, dystonia, teeth hypoplasia, poor communication skills, difficulty standing or walking, as well as developmental delay. Other names associated with SLC13A5 Epilepsy include SLC13A5 Citrate Transporter Disorder, Citrate Transporter Disorder, SLC13A5 Deficiency, Early Infantile Epilepsy Encephalopathy 25 (EIEE25), Developmental Epilepsy Encephalopathy 25 (DEE25), and Kohlschutter-Tonz Syndrome (non-ROGDI).
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
Isolated hyperchlorhidrosis, also known as Carbonic anhydrase XII deficiency, is a rare autosomal recessive genetic condition characterized by a lifelong tendency to lose massive amounts of sodium and chloride through sweat which leads to various symptoms.
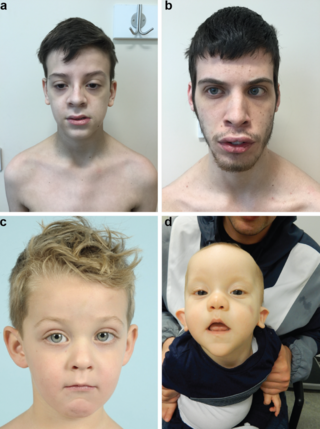
Beck–Fahrner syndrome, also known as BEFAHRS and TET3 deficiency, is a rare genetic disorder caused by mutations of the TET3 gene. The clinical presentation varies among individuals, but typically includes global developmental delay, slow progress in mental and physical activities, autism, decreased muscle tone, epilepsy and dysmorphic features.
