
Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.
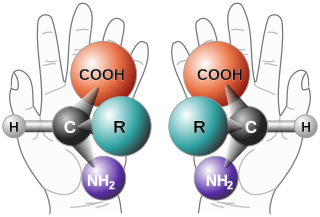
Enantioselective synthesis, also called asymmetric synthesis, is a form of chemical synthesis. It is defined by IUPAC as "a chemical reaction in which one or more new elements of chirality are formed in a substrate molecule and which produces the stereoisomeric products in unequal amounts."

Alfuzosin, sold under the brand name Uroxatral among others, is a medication of the α1 blocker class. It is used to treat benign prostatic hyperplasia (BPH).

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which an achiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes.
The Kiliani–Fischer synthesis, named for German chemists Heinrich Kiliani and Emil Fischer, is a method for synthesizing monosaccharides. It proceeds via synthesis and hydrolysis of a cyanohydrin, followed by reduction of the intermediate acid to the aldehyde, thus elongating the carbon chain of an aldose by one carbon atom while preserving stereochemistry on all the previously present chiral carbons. The new chiral carbon is produced with both stereochemistries, so the product of a Kiliani–Fischer synthesis is a mixture of two diastereomeric sugars, called epimers. For example, D-arabinose is converted to a mixture of D-glucose and D-mannose.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being awarded one half of the 2001 Nobel Prize in Chemistry.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
The Juliá–Colonna epoxidation is an asymmetric poly-leucine catalyzed nucleophilic epoxidation of electron deficient olefins in a triphasic system. The reaction was reported by Sebastian Juliá at the Chemical Institute of Sarriá in 1980, with further elaboration by both Juliá and Stefano Colonna.
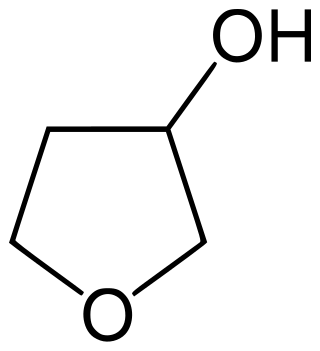
3-Hydroxytetrahydrofuran is a colorless liquid with a normal boiling point of 179 °C and boiling at 88−89 °C at 17 mmHg, with density. 3-OH THF is a useful pharmaceutical intermediate. The chiral version of this compound is an intermediate to launched retroviral drugs.
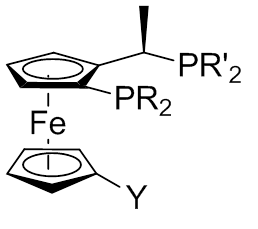
A Josiphos ligand is a type of chiral diphosphine which has been modified to be substrate-specific; they are widely used for enantioselective synthesis. They are named after the technician who made the first one, Josi Puleo.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. However, chemists have only recently attempted to harness the power of using hydrogen bonds to perform catalysis, and the field is relatively undeveloped compared to research in Lewis acid catalysis.
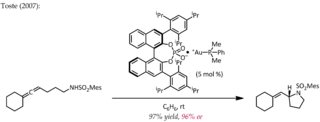
Asymmetric counteranion directed catalysis (ACDC) or chiral anion catalysis in enantioselective synthesis is the "induction of enantioselectivity in a reaction proceeding through a cationic intermediate by means of ion pairing with a chiral, enantiomerically pure anion provided by the catalyst". Although chiral Brønsted acid catalyzed reactions may well fall into this category of catalysis under the definition given here, the extent of proton transfer and the demarcation between hydrogen bonding and full proton transfer is often ambiguous. Hence, some authors may exclude ion pair formation by proton transfer as a type of chiral counteranion catalysis. The discussion below will focus on chiral ion pairs generated through means other than proton transfer.
In organic chemistry, the Keck asymmetric allylation is a chemical reaction that involves the nucleophilic addition of an allyl group to an aldehyde. The catalyst is a chiral complex that contains titanium as a Lewis acid. The chirality of the catalyst induces a stereoselective addition, so the secondary alcohol of the product has a predictable absolute stereochemistry based on the choice of catalyst. This name reaction is named for Gary Keck.
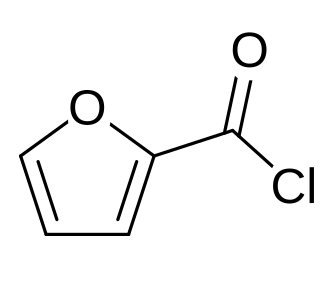
2-Furoyl chloride is an acyl chloride of furan. It takes the form of a corrosive liquid, which is more irritating to the eyes than benzoyl chloride. 2-Furoyl chloride is a useful pharmaceutical intermediate and is used in the synthesis of mometasone furoate, an antiinflammatory prodrug used in the treatment of skin disorders, hay fever and asthma.
