
A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. Antagonist drugs interfere in the natural operation of receptor proteins. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

Selective estrogen receptor modulators (SERMs), also known as estrogen receptor agonists/antagonists (ERAAs), are a class of drugs that act on estrogen receptors (ERs). Compared to pure ER agonists–antagonists, SERMs are more tissue-specific, allowing them to selectively inhibit or stimulate estrogen-like action in various tissues.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.

A selective progesterone receptor modulator (SPRM) is an agent that acts on the progesterone receptor (PR), the biological target of progestogens like progesterone. A characteristic that distinguishes such substances from full receptor agonists and full antagonists is that their action differs in different tissues, i.e. agonist in some tissues while antagonist in others. This mixed profile of action leads to stimulation or inhibition in tissue-specific manner, which further raises the possibility of dissociating undesirable adverse effects from the development of synthetic PR-modulator drug candidates.
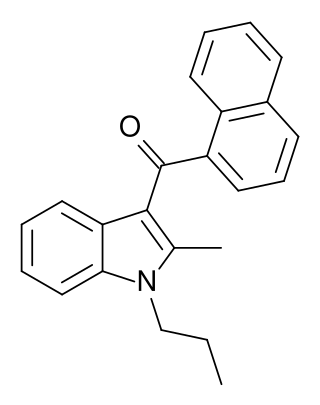
JWH-015 is a chemical from the naphthoylindole family that acts as a subtype-selective cannabinoid agonist. Its affinity for CB2 receptors is 13.8 nM, while its affinity for CB1 is 383 nM, meaning that it binds almost 28 times more strongly to CB2 than to CB1. However, it still displays some CB1 activity, and in some model systems can be very potent and efficacious at activating CB1 receptors, and therefore it is not as selective as newer drugs such as JWH-133. It has been shown to possess immunomodulatory effects, and CB2 agonists may be useful in the treatment of pain and inflammation. It was discovered and named after John W. Huffman.

Afimoxifene, also known as 4-hydroxytamoxifen (4-OHT) and by its tentative brand name TamoGel, is a selective estrogen receptor modulator (SERM) of the triphenylethylene group and an active metabolite of tamoxifen. The drug is under development under the tentative brand name TamoGel as a topical gel for the treatment of hyperplasia of the breast. It has completed a phase II clinical trial for cyclical mastalgia, but further studies are required before afimoxifene can be approved for this indication and marketed.

Xanomeline is a small molecule muscarinic acetylcholine receptor agonist that was first synthesized in a collaboration between Eli Lilly and Novo Nordisk as an investigational therapeutic being studied for the treatment of central nervous system (CNS) disorders.

Enobosarm, also formerly known as ostarine and by the developmental code names GTx-024, MK-2866, and S-22, is a selective androgen receptor modulator (SARM) which is under development for the treatment of androgen receptor-positive breast cancer in women and for improvement of body composition in people taking GLP-1 receptor agonists like semaglutide. It was also under development for a variety of other indications, including treatment of cachexia, Duchenne muscular dystrophy, muscle atrophy or sarcopenia, and stress urinary incontinence, but development for all other uses has been discontinued. Enobosarm was evaluated for the treatment of muscle wasting related to cancer in late-stage clinical trials, and the drug improved lean body mass in these trials, but it was not effective in improving muscle strength. As a result, enobosarm was not approved and development for this use was terminated. Enobosarm is taken by mouth.

Alazocine, also known more commonly as N-allylnormetazocine (NANM), is a synthetic opioid analgesic of the benzomorphan family related to metazocine, which was never marketed. In addition to its opioid activity, the drug is a sigma receptor agonist, and has been used widely in scientific research in studies of this receptor. Alazocine is described as a potent analgesic, psychotomimetic or hallucinogen, and opioid antagonist. Moreover, one of its enantiomers was the first compound that was found to selectively label the σ1 receptor, and led to the discovery and characterization of the receptor.

Selective glucocorticoid receptor modulators (SEGRMs) and selective glucocorticoid receptor agonists (SEGRAs) formerly known as dissociated glucocorticoid receptor agonists (DIGRAs) are a class of experimental drugs designed to share many of the desirable anti-inflammatory, immunosuppressive, or anticancer properties of classical glucocorticoid drugs but with fewer side effects such as skin atrophy. Although preclinical evidence on SEGRAMs’ anti-inflammatory effects are culminating, currently, the efficacy of these SEGRAMs on cancer are largely unknown.
In pharmacology and biochemistry, allosteric modulators are a group of substances that bind to a receptor to change that receptor's response to stimuli. Some of them, like benzodiazepines or alcohol, function as psychoactive drugs. The site that an allosteric modulator binds to is not the same one to which an endogenous agonist of the receptor would bind. Modulators and agonists can both be called receptor ligands.

Trimegestone, sold under the brand names Ondeva and Totelle among others, is a progestin medication which is used in menopausal hormone therapy and in the prevention of postmenopausal osteoporosis. It was also under development for use in birth control pills to prevent pregnancy, but ultimately was not marketed for this purpose. The medication is available alone or in combination with an estrogen. It is taken by mouth.
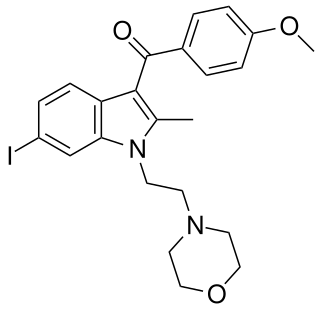
AM-630 (6-Iodopravadoline) is a drug that acts as a potent and selective inverse agonist for the cannabinoid receptor CB2, with a Ki of 32.1 nM at CB2 and 165x selectivity over CB1, at which it acted as a weak partial agonist. It is used in the study of CB2 mediated responses and has been used to investigate the possible role of CB2 receptors in the brain. AM-630 is significant as one of the first indole derived cannabinoid ligands substituted on the 6-position of the indole ring, a position that has subsequently been found to be important in determining affinity and efficacy at both the CB1 and CB2 receptors, and has led to the development of many related derivatives.
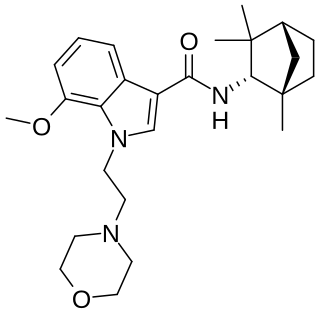
MN-25 (UR-12) is a drug invented by Bristol-Myers Squibb, that acts as a reasonably selective agonist of peripheral cannabinoid receptors. It has moderate affinity for CB2 receptors with a Ki of 11 nM, but 22x lower affinity for the psychoactive CB1 receptors with a Ki of 245 nM. The indole 2-methyl derivative has the ratio of affinities reversed however, with a Ki of 8 nM at CB1 and 29 nM at CB2, which contrasts with the usual trend of 2-methyl derivatives having increased selectivity for CB2 (cf. JWH-018 vs JWH-007, JWH-081 vs JWH-098).

Vosilasarm, also known by the development codes RAD140 and EP0062 and by the black-market name Testolone or Testalone, is a selective androgen receptor modulator (SARM) which is under development for the treatment of hormone-sensitive breast cancer. It is specifically under development for the treatment of androgen receptor-positive, estrogen receptor-negative, HER2-negative advanced breast cancer. Vosilasarm was also previously under development for the treatment of sarcopenia, osteoporosis, and weight loss due to cancer cachexia, but development for these indications was discontinued. The drug is taken by mouth.
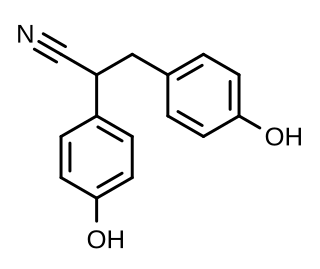
Diarylpropionitrile (DPN), also known as 2,3-bis(p-hydroxyphenyl)propionitrile (2,3-BHPPN), is a synthetic, nonsteroidal, and highly selective agonist of ERβ (IC50 = 15 nM) that is used widely in scientific research to study the function of this receptor. It is 70-fold more selective for ERβ over ERα, and has 100-fold lower affinity for GPER (GPR30) relative to estradiol. DPN produces antidepressant- and anxiolytic-like effects in animals via activation of the endogenous oxytocin system. First reported in 2001, DPN was the first selective ERβ agonist to be discovered, and was followed by prinaberel (ERB-041, WAY-202041), WAY-200070, and 8β-VE2 in 2004, ERB-196 (WAY-202196) in 2005, and certain phytoestrogens like liquiritigenin and nyasol (cis-hinokiresinol) since 2007.

LG121071 is a selective androgen receptor modulator (SARM) developed by Ligand Pharmaceuticals that was first described in 1999 and was the first orally active nonsteroidal androgen to be discovered. It is a tricyclic quinolone derivative, structurally distinct from other nonsteroidal AR agonists like andarine and enobosarm (ostarine). The drug acts as a high-affinity full agonist of the androgen receptor (AR), with a potency and efficacy that is said to be equivalent to that of dihydrotestosterone (DHT). Unlike testosterone, but similarly to DHT, LG121071 and other nonsteroidal androgens cannot be potentiated by 5α-reductase in androgenic tissues, and for this reason, show tissue-selective androgenic effects. In accordance, they are said to possess full anabolic activity with reduced androgenic activity, similarly to anabolic-androgenic steroids.

BMS-641988 is a nonsteroidal antiandrogen which was developed by Bristol-Myers Squibb for the treatment of prostate cancer but was never marketed. It acts as a potent competitive antagonist of the androgen receptor (AR) (Ki = 10 nM; IC50Tooltip half-maximal inhibitory concentration = 56 nM). The drug was found to have 20-fold higher affinity for the AR than bicalutamide in MDA-MB-453 cells, and showed 3- to 7-fold the antiandrogenic activity of bicalutamide in vitro. It may have some weak partial agonist activity at the androgen receptor. BMS-641988 is transformed by CYP3A4 into BMS-570511, and this metabolite is then reduced to BMS-501949 by cytosolic reductases. All three compounds show similar antiandrogenic activity. In addition to its antiandrogenic activity, BMS-641988 shows activity as a negative allosteric modulator of the GABAA receptor, and can produce seizures in animals at sufficiently high doses. It also shows some drug-induced QT prolongation. BMS-641988 reached phase I clinical trials prior to the discontinuation of its development. The clinical development of BMS-641988 was terminated due to the occurrence of a seizure in a patient during a phase I study.
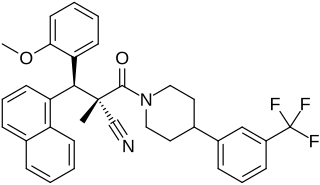
WAY-204688, also known as SIM-688, is a synthetic nonsteroidal estrogen and nuclear factor κB (NF-κB) inhibitor which was originated by ArQule and Wyeth and was under development by Wyeth for the treatment of rheumatoid arthritis, non-specific inflammation, and sepsis but was never marketed. It is a "pathway-selective" estrogen receptor (ER) ligand which inhibits NF-κB with an IC50Tooltip half-maximal inhibitory concentration of 122 nM and with maximal inhibition relative to estradiol of 94%. Inhibition of NF-κB by WAY-204688 appears to be dependent on agonism of the ERα, as it is reversed by the ERα antagonist fulvestrant, but is not dependent on the ERβ. In contrast to the case of NF-κB inhibition, WAY-204688 produces only slight elevation of creatine kinase in vitro, a measure of classical estradiol effects. It reached phase I clinical trials prior to the discontinuation of its development.

Tavapadon is a dopamine receptor agonist which is under development for the treatment of Parkinson's disease. It is under development by Cerevel Therapeutics, which acquired tavapadon from Pfizer in 2018. It is taken by mouth.
