Ataxia is a neurological sign consisting of lack of voluntary coordination of muscle movements that can include gait abnormality, speech changes, and abnormalities in eye movements. Ataxia is a clinical manifestation indicating dysfunction of parts of the nervous system that coordinate movement, such as the cerebellum. These nervous system dysfunctions occur in several different patterns, with different results and different possible causes. Ataxia can be limited to one side of the body, which is referred to as hemiataxia. Friedreich's ataxia has gait abnormality as the most commonly presented symptom. The word is from Greek α- [a negative prefix] + -τάξις [order] = "lack of order". Dystaxia is a mild degree of ataxia.
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.

Cerebellar hypoplasia is characterized by reduced cerebellar volume, even though cerebellar shape is (near) normal. It consists of a heterogeneous group of disorders of cerebellar maldevelopment presenting as early-onset non–progressive congenital ataxia, hypotonia and motor learning disability.
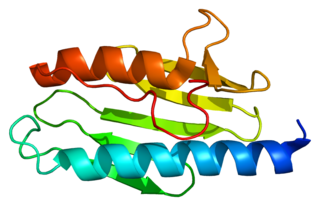
Friedreich's ataxia is an autosomal-recessive genetic disease that causes difficulty walking, a loss of sensation in the arms and legs, and impaired speech that worsens over time. Symptoms generally start between 5 and 20 years of age. Many develop hypertrophic cardiomyopathy and require a mobility aid such as a cane, walker, or wheelchair in their teens. As the disease progresses, some affected people lose their sight and hearing. Other complications may include scoliosis and diabetes mellitus.

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.
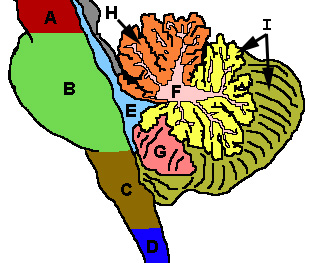
Cerebellar ataxia is a form of ataxia originating in the cerebellum. Non-progressive congenital ataxia (NPCA) is a classical presentation of cerebral ataxias.

Spinocerebellar ataxia type 13 (SCA13) is a rare autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, nystagmus, and ataxia of gait, stance and the limbs due to cerebellar dysfunction. Patients with SCA13 also tend to present with epilepsy, an inability to run, and increased reflexes. This cerebellar dysfunction is permanent and progressive. SCA13 is caused by mutations in KCNC3, a gene encoding a voltage-gated potassium channel KV3.3. There are two known mutations in this gene causative for SCA13. Unlike many other types of SCA, these are not polyglutamine expansions but, rather, point mutations resulting in channels with no current or altered kinetics.
Frataxin is a protein that in humans is encoded by the FXN gene.
Ramsay Hunt syndrome type 1 is a rare, degenerative, neurological disorder characterized by myoclonus epilepsy, intention tremor, progressive ataxia and occasionally cognitive impairment
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Peter Kynaston "PK" Thomas was a Welsh academic neurologist, author, teacher and administrator. From 1974 to 1991 he was Professor of Neurology at the University of London. He was a fellow of University College London and the Royal Society of Medicine, and was the recipient of the Medal of the Association of British Neurologists. Thomas was, at various times, the president of the Association of British Neurologists, the European Neurological Society, and the Peripheral Nerve Society.

Autosomal recessive cerebellar ataxia type 1 (ARCA1) is a condition characterized by progressive problems with movement. Signs and symptoms of the disorder first appear in early to mid-adulthood. People with this condition initially experience impaired speech (dysarthria), problems with coordination and balance (ataxia), or both. They may also have difficulty with movements that involve judging distance or scale (dysmetria). Other features of ARCA1 include abnormal eye movements (nystagmus) and problems following the movements of objects with their eyes. The movement problems are slowly progressive, often resulting in the need for a cane, walker, or wheelchair.
Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.
Non-progressive congenital ataxia (NPCA) is a non-progressive form of cerebellar ataxia which can occur with or without cerebellar hypoplasia.
Autosomal recessive cerebellar ataxia describes a heterogeneous group of rare genetic disorders with an autosomal recessive inheritance pattern and a clinical phenotype involving cerebellar ataxia.
COACH syndrome, also known as Joubert syndrome with hepatic defect, is a rare autosomal recessive genetic disease. The name is an acronym of the defining signs: cerebellar vermis aplasia, oligophrenia, congenital ataxia, coloboma and hepatic fibrosis. The condition is associated with moderate intellectual disability. It falls under the category of a Joubart Syndrome-related disorder (JSRD).
William Walton Gooddy (1916–2004) was an English neurologist.
Neck-tongue syndrome (NTS), which was first recorded in 1980, is a rare disorder characterized by neck pain with or without tingling and numbness of the tongue on the same side as the neck pain. Sharp lateral movement of the head triggers the pain, usually lasting from a few seconds to a few minutes.Headaches may occur with the onset of NTS. The typical age of onset is around adolescence and may occur as early as 8–15 years old. However, it is worth noting that clinical onset can occur earlier or later and NTS onset related to trauma can occur at any age, beginning after the incident.
Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS) is an autosomal recessive late-onset heredodegenerative multisystem neurological disease. The symptoms include poor balance and difficulty walking. Chronic cough and difficulty swallowing may also be present. Clinical findings include ataxia, sensory neuropathy, and absence of the vestibulo–ocular reflex. The syndrome was initially described in 2004. In 2019, the cause was identified as biallelic pentanucleotide expansion in the RFC1 gene.

Boucher-Neuhäuser syndrome is a very rare genetic disorder which is characterized by a triad consisting of cerebellar ataxia, chorioretinal dystrophy, and hypogonadism.
