Related Research Articles

Chorea-acanthocytosis is a rare hereditary disease caused by a mutation in a gene that directs structural proteins in red blood cells. It belongs to a group of four diseases characterized under the name neuroacanthocytosis. When a patient's blood is viewed under a microscope, some of the red blood cells appear thorny. These thorny cells are called acanthocytes.

Neuronal ceroid lipofuscinosis is the general name for a family of at least eight genetically separate neurodegenerative lysosomal storage diseases that result from excessive accumulation of lipopigments (lipofuscin) in the body's tissues. These lipopigments are made up of fats and proteins. Their name comes from the word stem "lipo-", which is a variation on lipid, and from the term "pigment", used because the substances take on a greenish-yellow color when viewed under an ultraviolet light microscope. These lipofuscin materials build up in neuronal cells and many organs, including the liver, spleen, myocardium, and kidneys.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden–Spatz syndrome, is a genetic degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. Neurodegeneration in PKAN is accompanied by an excess of iron that progressively builds up in the brain.

Aceruloplasminemia is a rare autosomal recessive disorder in which the liver can not synthesize the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time. .
Neuroacanthocytosis is a label applied to several genetic neurological conditions in which the blood contains misshapen, spiculated red blood cells called acanthocytes.
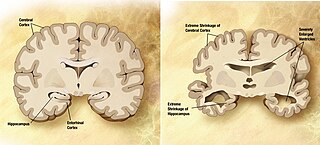
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.
Glucose transporter 1, also known as solute carrier family 2, facilitated glucose transporter member 1 (SLC2A1), is a uniporter protein that in humans is encoded by the SLC2A1 gene. GLUT1 facilitates the transport of glucose across the plasma membranes of mammalian cells. This gene encodes a facilitative glucose transporter that is highly expressed in erythrocytes and endothelial cells, including cells of the blood–brain barrier. The encoded protein is found primarily in the cell membrane and on the cell surface, where it can also function as a receptor for human T-cell leukemia virus (HTLV) I and II. GLUT1 accounts for 2 percent of the protein in the plasma membrane of erythrocytes.
Infantile neuroaxonal dystrophy is a rare pervasive developmental disorder that primarily affects the nervous system. Individuals with infantile neuroaxonal dystrophy typically do not have any symptoms at birth, but between the ages of about 6 and 18 months they begin to experience delays in acquiring new motor and intellectual skills, such as crawling or beginning to speak. Eventually they lose previously acquired skills.

85 kDa calcium-independent phospholipase A2, also known as 85/88 kDa calcium-independent phospholipase A2, Group VI phospholipase A2, Intracellular membrane-associated calcium-independent phospholipase A2 beta, or Patatin-like phospholipase domain-containing protein 9 is an enzyme that in humans is encoded by the PLA2G6 gene.

Mitochondrial import inner membrane translocase subunit Tim8 A, also known as deafness-dystonia peptide or protein is an enzyme that in humans is encoded by the TIMM8A gene. This translocase has similarity to yeast mitochondrial proteins that are involved in the import of metabolite transporters from the cytoplasm into the mitochondrial inner membrane. The gene is mutated in deafness-dystonia syndrome and it is postulated that MTS/DFN-1 is a mitochondrial disease caused by a defective mitochondrial protein import system.

Vacuolar protein sorting ortholog 35 (VPS35) is a protein involved in autophagy and is implicated in neurodegenerative diseases, such as Parkinson's disease (PD) and Alzheimer's disease (AD). VPS35 is part of a complex called the retromer, which is responsible for transporting select cargo proteins between vesicular structures and the Golgi apparatus. Mutations in the VPS35 gene (VPS35) cause aberrant autophagy, where cargo proteins fail to be transported and dysfunctional or unnecessary proteins fail to be degraded. There are numerous pathways affected by altered VPS35 levels and activity, which have clinical significance in neurodegeneration. There is therapeutic relevance for VPS35, as interventions aimed at correcting VPS35 function are in speculation.
Woodhouse–Sakati syndrome, is a rare autosomal recessive multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system.
WD repeat domain phosphoinositide-interacting protein 4 (WIPI-4) is a protein that in humans is encoded by the WDR45 gene. Mutations in this gene cause a distinct form of Neurodegeneration with brain iron accumulation (NBIA) called Beta-propeller protein-associated neurodegeneration (BPAN).

Basal ganglia disease is a group of physical problems that occur when the group of nuclei in the brain known as the basal ganglia fail to properly suppress unwanted movements or to properly prime upper motor neuron circuits to initiate motor function. Research indicates that increased output of the basal ganglia inhibits thalamocortical projection neurons. Proper activation or deactivation of these neurons is an integral component for proper movement. If something causes too much basal ganglia output, then the ventral anterior (VA) and ventral lateral (VL) thalamocortical projection neurons become too inhibited, and one cannot initiate voluntary movement. These disorders are known as hypokinetic disorders. However, a disorder leading to abnormally low output of the basal ganglia leads to reduced inhibition, and thus excitation, of the thalamocortical projection neurons which synapse onto the cortex. This situation leads to an inability to suppress unwanted movements. These disorders are known as hyperkinetic disorders.

Neuroferritinopathy is a genetic neurodegenerative disorder characterized by the accumulation of iron in the basal ganglia, cerebellum, and motor cortex of the human brain. Symptoms, which are extrapyramidal in nature, progress slowly and generally do not become apparent until adulthood. These symptoms include chorea, dystonia, and cognitive deficits which worsen with age.

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive dystonia or ataxia, visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.
Neurodegeneration with brain iron accumulation is a heterogenous group of inherited neurodegenerative diseases, still under research, in which iron accumulates in the basal ganglia, either resulting in progressive dystonia, parkinsonism, spasticity, optic atrophy, retinal degeneration, neuropsychiatric, or diverse neurologic abnormalities. Some of the NBIA disorders have also been associated with several genes in synapse and lipid metabolism related pathways. NBIA is not one disease but an entire group of disorders, characterized by an accumulation of brain iron, sometimes in the presence of axonal spheroids in the central nervous system.

Solute carrier family 25 member 46 is a protein that in humans is encoded by the SLC25A46 gene. This protein is a member of the SLC25 mitochondrial solute carrier family. It is a transmembrane protein located in the mitochondrial outer membrane involved in lipid transfer from the endoplasmic reticulum (ER) to mitochondria. Mutations in this gene result in neuropathy and optic atrophy.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 Gregory A, Hartig M, Prokisch H, Kmiec T, Hogarth P, Hayflick SJ (2014). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). "Mitochondrial Membrane Protein-Associated Neurodegeneration". GeneReviews [Internet]. PMID 24575447.
- 1 2 3 4 "Mitochondrial membrane protein-associated neurodegeneration: MedlinePlus Genetics". medlineplus.gov. Retrieved 21 April 2022.
- 1 2 3 4 Hogarth, P (January 2015). "Neurodegeneration with Brain Iron Accumulation: Diagnosis and Management". Journal of Movement Disorders. 8 (1): 1–13. doi:10.14802/jmd.14034. PMC 4298713 . PMID 25614780.
- 1 2 Online Mendelian Inheritance in Man (OMIM): NEURODEGENERATION WITH BRAIN IRON ACCUMULATION 4 - 614298
- ↑ Online Mendelian Inheritance in Man (OMIM): CHROMOSOME 19 OPEN READING FRAME 12 - 614297
- 1 2 Spaull, R; Soo, A; Hogarth; Hayflick, S; Kurian, M (November 2021). "Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation". Tremor and Other Hyperkinetic Movements. 11 (1): 51. doi: 10.5334/tohm.661 . ISSN 2160-8288. PMC 8641530 . PMID 34909266.
- ↑ Hartig, Monika B.; Iuso, Arcangela; Haack, Tobias; Kmiec, Tomasz; Jurkiewicz, Elzbieta; Heim, Katharina; Roeber, Sigrun; Tarabin, Victoria; Dusi, Sabrina; Krajewska-Walasek, Malgorzata; Jozwiak, Sergiusz (October 2011). "Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation". The American Journal of Human Genetics. 89 (4): 543–550. doi:10.1016/j.ajhg.2011.09.007. PMC 3188837 . PMID 21981780.
- ↑ Hogarth, P.; Gregory, A.; Kruer, M. C.; Sanford, L.; Wagoner, W.; Natowicz, M. R.; Egel, R. T.; Subramony, S. H.; Goldman, J. G.; Berry-Kravis, E.; Foulds, N. C. (15 January 2013). "New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN". Neurology. 80 (3): 268–275. doi:10.1212/WNL.0b013e31827e07be. ISSN 0028-3878. PMC 3589182 . PMID 23269600.