
Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders in the current classification, with the latest type discovered in 2018. Symptoms include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily.
Dystonia is a neurological hyperkinetic movement disorder in which sustained or repetitive muscle contractions result in twisting and repetitive movements or abnormal fixed postures. The movements may resemble a tremor. Dystonia is often intensified or exacerbated by physical activity, and symptoms may progress into adjacent muscles.

Chorea-acanthocytosis is a rare hereditary disease caused by a mutation in a gene that directs structural proteins in red blood cells. It belongs to a group of four diseases characterized under the name neuroacanthocytosis. When a patient's blood is viewed under a microscope, some of the red blood cells appear thorny. These thorny cells are called acanthocytes.
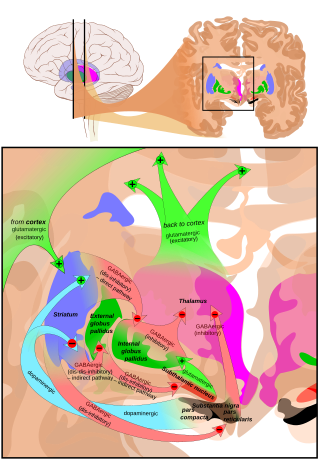
Hyperkinesia refers to an increase in muscular activity that can result in excessive abnormal movements, excessive normal movements, or a combination of both. Hyperkinesia is a state of excessive restlessness which is featured in a large variety of disorders that affect the ability to control motor movement, such as Huntington's disease. It is the opposite of hypokinesia, which refers to decreased bodily movement, as commonly manifested in Parkinson's disease.

Leukodystrophies are a group of, usually, inherited disorders, characterized by degeneration of the white matter in the brain. The word leukodystrophy comes from the Greek roots leuko, "white", dys, "abnormal" and troph, "growth". The leukodystrophies are caused by imperfect growth or development of the glial cells which produce the myelin sheath, the fatty insulating covering around nerve fibers. Leukodystrophies may be classified as hypomyelinating or demyelinating diseases, respectively, depending on whether the damage is present before birth or occurs after. Other demyelinating diseases are usually not congenital and have a toxic or autoimmune cause.
Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B. These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues, and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death. The rare autosomal recessive neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.

Blepharospasm is any abnormal contraction of the orbicularis oculi muscle. The condition should be distinguished from the more common, and milder, involuntary quivering of an eyelid, known as myokymia, or fasciculation. In most cases, blepharospasm symptoms last for a few days and then disappear without treatment, but in some cases the twitching is chronic and persistent, causing life-long challenges. In these cases, the symptoms are often severe enough to result in functional blindness. The person's eyelids feel like they are clamping shut and will not open without great effort. People have normal eyes, but for periods of time are effectively blind due to their inability to open their eyelids. In contrast, the reflex blepharospasm is due to any pain in and around the eye.

Spasmodic torticollis is an extremely painful chronic neurological movement disorder causing the neck to involuntarily turn to the left, right, upwards, and/or downwards. The condition is also referred to as "cervical dystonia". Both agonist and antagonist muscles contract simultaneously during dystonic movement. Causes of the disorder are predominantly idiopathic. A small number of patients develop the disorder as a result of another disorder or disease. Most patients first experience symptoms midlife. The most common treatment for spasmodic torticollis is the use of botulinum toxin type A.
Spasmodic dysphonia, also known as laryngeal dystonia, is a disorder in which the muscles that generate a person's voice go into periods of spasm. This results in breaks or interruptions in the voice, often every few sentences, which can make a person difficult to understand. The person's voice may also sound strained or they may be nearly unable to speak. Onset is often gradual and the condition is lifelong.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).

Torsin-1A (TorA) also known as dystonia 1 protein (DYT1) is a protein that in humans is encoded by the TOR1A gene. TorA localizes to the endoplasmic reticulum and contiguous perinuclear space, where its ATPase activity is activated by either LULL1 or LAP1, respectively.
Torsin-2A is a protein that in humans is encoded by the TOR2A gene.
Myoclonic dystonia or Myoclonus dystonia syndrome is a rare movement disorder that induces spontaneous muscle contraction causing abnormal posture. The prevalence of myoclonus dystonia has not been reported, however, this disorder falls under the umbrella of movement disorders which affect thousands worldwide. Myoclonus dystonia results from mutations in the SGCE gene coding for an integral membrane protein found in both neurons and muscle fibers. Those suffering from this disease exhibit symptoms of rapid, jerky movements of the upper limbs (myoclonus), as well as distortion of the body's orientation due to simultaneous activation of agonist and antagonist muscles (dystonia).

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive or , visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.

Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.
X-linked dystonia parkinsonism (XDP), also known as Lubag Syndrome or X-linked Dystonia of Panay, is a rare x-linked progressive movement disorder with high penetrance found almost exclusively in males from Panay, Philippines. It is characterized by dystonic movements first typically occurring in the 3rd and 4th decade of life. The dystonic movements often either coexist or develop into parkinsonism within 10 years of disease onset.
Tyrosine hydroxylase deficiency (THD) is a disorder caused by disfunction of tyrosine hydroxylase, an enzyme involved in the biosynthesis of dopamine. This condition is one of the causes of dopa-responsive dystonia.
Spastic paraplegia 15 (SPG15) is a form of hereditary spastic paraplegia that commonly becomes apparent during childhood or adolescence. The disease is caused by mutations within the ZFYVE26 gene - also known as the SPG15 gene - and is passed down in an autosomal recessive manner.