
.NET Bio is an open source bioinformatics and genomics library created to enable simple loading, saving and analysis of biological data. It was designed for .NET Standard 2.0 and was part of the Microsoft Biology Initiative in the eScience division.
.NET Bio is an open source bioinformatics and genomics library created to enable simple loading, saving and analysis of biological data. It was designed for .NET Standard 2.0 and was part of the Microsoft Biology Initiative in the eScience division.
.NET Bio was originally built and released by Microsoft Research under the name Microsoft Biology Foundation (MBF) and was later repackaged and released by the Outercurve Foundation [1] as a fully public and open source project under the Apache License 2.0. [2]
The library consists of a set of object-oriented classes written in C# to perform common bioinformatic tasks such as:
Even though the library itself is written in C#, it may be used from any .NET compatible language and has samples of various usages including from IronPython scripting. [4]

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
F# is a general-purpose, strongly typed, multi-paradigm programming language that encompasses functional, imperative, and object-oriented programming methods. It is most often used as a cross-platform Common Language Infrastructure (CLI) language on .NET, but can also generate JavaScript and graphics processing unit (GPU) code.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
In bioinformatics, sequence clustering algorithms attempt to group biological sequences that are somehow related. The sequences can be either of genomic, "transcriptomic" (ESTs) or protein origin. For proteins, homologous sequences are typically grouped into families. For EST data, clustering is important to group sequences originating from the same gene before the ESTs are assembled to reconstruct the original mRNA.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a huge range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.
The Biopython project is an open-source collection of non-commercial Python tools for computational biology and bioinformatics, created by an international association of developers. It contains classes to represent biological sequences and sequence annotations, and it is able to read and write to a variety of file formats. It also allows for a programmatic means of accessing online databases of biological information, such as those at NCBI. Separate modules extend Biopython's capabilities to sequence alignment, protein structure, population genetics, phylogenetics, sequence motifs, and machine learning. Biopython is one of a number of Bio* projects designed to reduce code duplication in computational biology.

The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings of nucleic acid sequences or protein sequences. Instead of looking at the entire sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure.
Bioconductor is a free, open source and open development software project for the analysis and comprehension of genomic data generated by wet lab experiments in molecular biology.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
BioPHP is a collection of open-source PHP code, with classes for DNA and protein sequence analysis, alignment, database parsing, and other bioinformatics tools. BioRuby is released under the GNU GPL version 2 licence and is one of a number of Bio* projects, designed to reduce code duplication. As an open source bioinformatics project, BioPHP is affiliated with the Open Bioinformatics Foundation.
UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.

RNA-Seq is a technique that uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
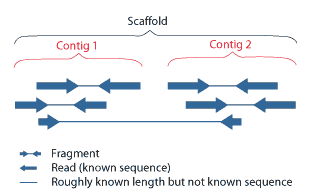
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
De novo sequence assemblers are a type of program that assembles short nucleotide sequences into longer ones without the use of a reference genome. These are most commonly used in bioinformatic studies to assemble genomes or transcriptomes. Two common types of de novo assemblers are greedy algorithm assemblers and De Bruijn graph assemblers.
Microsoft, a technology company historically known for its opposition to the open source software paradigm, turned to embrace the approach in the 2010s. From the 1970s through 2000s under CEOs Bill Gates and Steve Ballmer, Microsoft viewed the community creation and sharing of communal code, later to be known as free and open source software, as a threat to its business, and both executives spoke negatively against it. In the 2010s, as the industry turned towards cloud, embedded, and mobile computing—technologies powered by open source advances—CEO Satya Nadella led Microsoft towards open source adoption although Microsoft's traditional Windows business continued to grow throughout this period generating revenues of 26.8 billion in the third quarter of 2018, while Microsoft's Azure cloud revenues nearly doubled.
| Main projects |
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MSR Labs applied research |
| ||||||||||||||