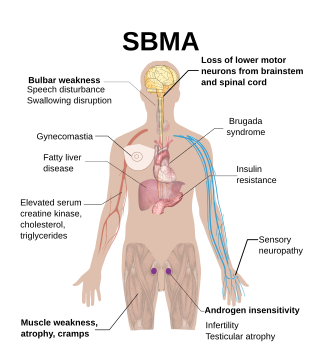
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.
In genetics, trinucleotide repeat disorders, a subset of microsatellite expansion diseases, are a set of over 30 genetic disorders caused by trinucleotide repeat expansion, a kind of mutation in which repeats of three nucleotides increase in copy numbers until they cross a threshold above which they cause developmental, neurological or neuromuscular disorders. In addition to the expansions of these trinucleotide repeats, expansions of one tetranucleotide (CCTG), five pentanucleotide, three hexanucleotide, and one dodecanucleotide (CCCCGCCCCGCG) repeat cause 13 other diseases. Depending on its location, the unstable trinucleotide repeat may cause defects in a protein encoded by a gene; change the regulation of gene expression; produce a toxic RNA, or lead to production of a toxic protein. In general, the larger the expansion the faster the onset of disease, and the more severe the disease becomes.

Huntingtin(Htt) is the protein coded for in humans by the HTT gene, also known as the IT15 ("interesting transcript 15") gene. Mutated HTT is the cause of Huntington's disease (HD), and has been investigated for this role and also for its involvement in long-term memory storage.
A trinucleotide repeat expansion, also known as a triplet repeat expansion, is the DNA mutation responsible for causing any type of disorder categorized as a trinucleotide repeat disorder. These are labelled in dynamical genetics as dynamic mutations. Triplet expansion is caused by slippage during DNA replication, also known as "copy choice" DNA replication. Due to the repetitive nature of the DNA sequence in these regions, 'loop out' structures may form during DNA replication while maintaining complementary base pairing between the parent strand and daughter strand being synthesized. If the loop out structure is formed from the sequence on the daughter strand this will result in an increase in the number of repeats. However, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. It appears that expansion of these repeats is more common than reduction. Generally, the larger the expansion the more likely they are to cause disease or increase the severity of disease. Other proposed mechanisms for expansion and reduction involve the interaction of RNA and DNA molecules.

Ataxin-1 is a DNA-binding protein which in humans is encoded by the ATXN1 gene.
Ataxin 7 (ATXN7) is a protein of the SCA7 gene, which contains 892 amino acids with an expandable poly(Q) region close to the N-terminus. The expandable poly(Q) motif region in the protein contributes crucially to spinocerebellar ataxia (SCA) pathogenesis by the induction of intranuclear inclusion bodies. ATXN7 is associated with both olivopontocerebellar atrophy type 3 (OPCA3) and spinocerebellar ataxia type 7 (SCA7).

Survival of motor neuron 1 (SMN1), also known as component of gems 1 or GEMIN1, is a gene that encodes the SMN protein in humans.

Ataxin-2 is a protein that in humans is encoded by the ATXN2 gene. Mutations in ATXN2 cause spinocerebellar ataxia type 2 (SCA2).

Cav2.1, also called the P/Q voltage-dependent calcium channel, is a calcium channel found mainly in the brain. Specifically, it is found on the presynaptic terminals of neurons in the brain and cerebellum. Cav2.1 plays an important role in controlling the release of neurotransmitters between neurons. It is composed of multiple subunits, including alpha-1, beta, alpha-2/delta, and gamma subunits. The alpha-1 subunit is the pore-forming subunit, meaning that the calcium ions flow through it. Different kinds of calcium channels have different isoforms (versions) of the alpha-1 subunit. Cav2.1 has the alpha-1A subunit, which is encoded by the CACNA1A gene. Mutations in CACNA1A have been associated with various neurologic disorders, including familial hemiplegic migraine, episodic ataxia type 2, and spinocerebellar ataxia type 6.

Ataxin-3 is a protein that in humans is encoded by the ATXN3 gene.

Brain-specific angiogenesis inhibitor 1-associated protein 2 is a protein that in humans is encoded by the BAIAP2 gene.

Polyglutamine-binding protein 1 (PQBP1) is a protein that in humans is encoded by the PQBP1 gene.

Membrane-associated guanylate kinase, WW and PDZ domain-containing protein 1 is an enzyme that in humans is encoded by the MAGI1 gene.

Arginine-glutamic acid dipeptide repeats protein is a protein that in humans is encoded by the RERE gene.

Membrane-associated guanylate kinase, WW and PDZ domain-containing protein 2 also known as membrane-associated guanylate kinase inverted 2 (MAGI-2) and atrophin-1-interacting protein 1 (AIP-1) is an enzyme that in humans is encoded by the MAGI2 gene.

Junctophilin-3 (JPH3) is a protein residing in humans that is encoded by the JPH3 gene. The gene is approximately 97 kilobases long and is located at chromosomal position 16q24.2. Junctophilin proteins are associated with the formation of junctional membrane complexes, which link the plasma membrane with the endoplasmic reticulum in excitable cells. JPH3 is localized to the brain and is associated with motor coordination and memory neurons.
Ataxin 8 opposite strand, also known as ATXN8OS, is a human gene.

Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.

Mild androgen insensitivity syndrome (MAIS) is a condition that results in a mild impairment of the cell's ability to respond to androgens. The degree of impairment is sufficient to impair spermatogenesis and / or the development of secondary sexual characteristics at puberty in males, but does not affect genital differentiation or development. Female genital and sexual development is not significantly affected by the insensitivity to androgens; as such, MAIS is only diagnosed in males. The clinical phenotype associated with MAIS is a normal male habitus with mild spermatogenic defect and / or reduced secondary terminal hair.
Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.






