Related Research Articles

The Waste Electrical and Electronic Equipment Directive is a European Community Directive, numbered 2012/19/EU, concerned with waste electrical and electronic equipment (WEEE). Together with the RoHS Directive 2011/65/EU, it became European Law in February 2003. The WEEE Directive set collection, recycling and recovery targets for all types of electrical goods, with a minimum rate of 4 kilograms (9 lb) per head of population per annum recovered for recycling by 2009. The RoHS Directive set restrictions upon European manufacturers as to the material content of new electronic equipment placed on the market.

The Restriction of Hazardous Substances Directive 2002/95/EC, short for Directive on the restriction of the use of certain hazardous substances in electrical and electronic equipment, was adopted in February 2003 by the European Union.

The regulation of therapeutic goods, defined as drugs and therapeutic devices, varies by jurisdiction. In some countries, such as the United States, they are regulated at the national level by a single agency. In other jurisdictions they are regulated at the state level, or at both state and national levels by various bodies, as in Australia.

The presence of the logo on commercial products indicates that the manufacturer or importer affirms the goods' conformity with European health, safety, and environmental protection standards. It is not a quality indicator or a certification mark. The CE marking is required for goods sold in the European Economic Area (EEA); goods sold elsewhere may also carry the mark.

A medical device is any device intended to be used for medical purposes. Significant potential for hazards are inherent when using a device for medical purposes and thus medical devices must be proved safe and effective with reasonable assurance before regulating governments allow marketing of the device in their country. As a general rule, as the associated risk of the device increases the amount of testing required to establish safety and efficacy also increases. Further, as associated risk increases the potential benefit to the patient must also increase.
Type approval or certificate of conformity is granted to a product that meets a minimum set of regulatory, technical and safety requirements. Generally, type approval is required before a product is allowed to be sold in a particular country, so the requirements for a given product will vary around the world. Processes and certifications known as type approval in English are often called homologation, or some cognate expression, in other European languages.
ISO 13485Medical devices -- Quality management systems -- Requirements for regulatory purposes is a voluntary standard, published by International Organization for Standardization (ISO) for the first time in 1996, and contains a comprehensive quality management system for the design and manufacture of medical devices. The latest version of this standard supersedes earlier documents such as EN 46001 and EN 46002 (1996), the previously published ISO 13485, and ISO 13488.
The Good Clinical Practice Directive lays down principles and detailed guidelines for good clinical practice as regards conducting clinical trials of medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products.
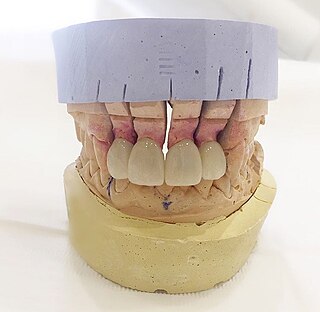
Dental laboratories manufacture or customize a variety of products to assist in the provision of oral health care by a licensed dentist. These products include crowns, bridges, dentures and other dental products. Dental lab technicians follow a prescription from a licensed dentist when manufacturing these items, which include prosthetic devices and therapeutic devices. The FDA regulates these products as medical devices and they are therefore subject to FDA's good manufacturing practice ("GMP") and quality system ("QS") requirements. In most cases, however, they are exempt from manufacturer registration requirements. Some of the most common restorations manufactured include crowns, bridges, dentures, and dental implants. Dental implants is one of the most advanced dental technologies in the field of dentistry.
The Medical Device Directive—Council Directive 93/42/EEC of 14 June 1993 concerning medical devices—is intended to harmonise the laws relating to medical devices within the European Union. The MD Directive is a 'New Approach' Directive and consequently in order for a manufacturer to legally place a medical device on the European market the requirements of the MD Directive have to be met. Manufacturers' products meeting 'harmonised standards' have a presumption of conformity to the Directive. Products conforming with the MD Directive must have a CE mark applied. The Directive was most recently reviewed and amended by the 2007/47/EC and a number of changes were made. Compliance with the revised directive became mandatory on 21 March 2010.
A European Authorised Representative (E.A.R.) serves as a legal entity designated by non European Union (EU) manufacturers, to represent them in the EU and ensure their compliance with the European Directives. The CE certificate and declaration of conformity can only be issued by a company located in the European Union.
The Italian Ministry of Health (MOH) has implemented mandatory procedures for the Italian registration of medical devices as of 1 May 2007.
Council Directive 76/768/EEC of 27 July 1976 on the approximation of the laws of the Member States relating to cosmetic products was the main European Union law on the safety of cosmetics. It was made under Art. 100 of the Treaty of Rome. By agreement, it was also applicable in the European Economic Area.
Design controls designates the application of a formal methodology to the conduct of product development activities. It is often mandatory to implement such practice when designing and developing products within regulated industries.
ISO 14971Medical devices — Application of risk management to medical devices is a voluntary standard for the application of risk management to medical devices. "Voluntary standards do not replace national laws, with which standards' users are understood to comply and which take precedence" over voluntary standards such as ISO 13485 and ISO 14971. The ISO Technical Committee responsible for the maintenance of this standard is ISO/ TC 210 working with IEC/SC62A through Joint Working Group one (JWG1). This standard is the culmination of the work starting in ISO/IEC Guide 51, and ISO/IEC Guide 63. The third edition of ISO 14971 was published in December 2019 and supersedes the second edition of ISO 14971.
The Machinery Directive, Directive 2006/42/EC of the European Parliament and of the Council of 17 May 2006 is a European Union directive concerning machinery and certain parts of machinery. Its main intent is to ensure a common safety level in machinery placed on the market or put in service in all member states and to ensure freedom of movement within the European Union by stating that "member states shall not prohibit, restrict or impede the placing on the market and/or putting into service in their territory of machinery which complies with [the] Directive".

European company law is the part of European Union law which concerns the formation, operation and insolvency of companies in the European Union. The EU creates minimum standards for companies throughout the EU, and has its own corporate forms. All member states continue to operate separate companies acts, which are amended from time to time to comply with EU Directives and Regulations. There is, however, also the option of businesses to incorporate as a Societas Europaea (SE), which allows a company to operate across all member states.
Regulation (EU) 2017/745 is a regulation of the European Union on the clinical investigation and sale of medical devices for human use. It repeals Directive 93/42/EEC (MDD), which concerns medical devices, and Directive 90/385/EEC, which concerns active implantable medical devices, on 26 May 2021.
Regulation (EU) 2017/746 (IVDR) is a regulation of the European Union on the placing on the market and putting into service of in vitro diagnostic medical devices (IVD), repealing Directive 98/79/EC (IVDD), which also concerned IVD. The regulation was published in April 2017 and is closely aligned to the EU regulation on medical devices. Changes compared to the IVDD include changes in device classification, stricter oversight of manufacturers by Notified Bodies, introduction of the "Person Responsible for Regulatory Compliance" (PRRC), the requirement of UDI marking for devices, common specifications, Eudamed registration, and increased post-market surveillance activities.
References
- ↑ Therapeutic Goods (Medical Devices) Regulations 2002 (Statutory Rules No. 236, 2002) (Regulations). Australia. 14 January 2022.
- 1 2 Medical Devices Regulations SOR/98-282 (PDF) (Regulations). 7 May 1998.
- ↑ Guidance for Health Care Professionals on Special Access and Custom-Made Medical Devices. Ottawa, Ontario, Canada: Health Canada. 18 February 2016.
- 1 2 European Parliament / European Council. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (Regulation (EU) 2017/745, Article 2). The European Parliament and Council of the European Union. 5 April 2017. p. 16.
- 1 2 The Medical Devices Regulations 2002 (Statutory Instrument 2002/618) (PDF). United Kingdom. 20 May 2002.
- ↑ Federal Food, Drug, and Cosmetic Act (PDF) (Section 520(b)). 11 December 2020. p. 425–426.
- ↑ "Custom-made devices in Great Britain". UK Government. Medicines and Healthcare products Regulatory Agency. 31 December 2020. Retrieved 20 January 2022.
- ↑ "Custom-made medical devices: Information for sponsors, health professionals & manufacturers" (PDF). Australian Government. Therapeutic Goods Administration. November 2021. Retrieved 20 January 2022.
- ↑ Council Directive 93/42/EEC of 14 June 1993 [1] concerning medical devices, OJ No L 169/1 of 1993-07-12 (Article 2). Council of the European Union (Medical Device Directive). 14 June 1993.
- ↑ Green, James I. J. (15 March 2021). "Medical Device Regulation: Requirements for Dental Professionals Who Prescribe and Manufacture Custom-Made Devices". Primary Dental Journal. 10 (1): 64–88. doi:10.1177/2050168420980980. PMID 33722134. S2CID 232245011.
- ↑ The Medical Devices (Amendment etc.) (EU Exit) Regulations 2019 (Statutory Instrument 2019/791) (PDF). United Kingdom. 1 April 2019.
- ↑ The Medical Devices (Amendment etc.) (EU Exit) Regulations 2020 (Statutory Instrument 2020/1478) (PDF). United Kingdom. 8 December 2020.
- ↑ PROTOCOL ON IRELAND/NORTHERN IRELAND (PDF). United Kingdom. 17 October 2019.
- ↑ Custom Device Exemption: Guidance for Industry and Food and Drug Administration Staff. Washington, D.C., United States: Federal government of the United States. 14 January 2014.