
Microscopy is the technical field of using microscopes to view objects and areas of objects that cannot be seen with the naked eye. There are three well-known branches of microscopy: optical, electron, and scanning probe microscopy, along with the emerging field of X-ray microscopy.
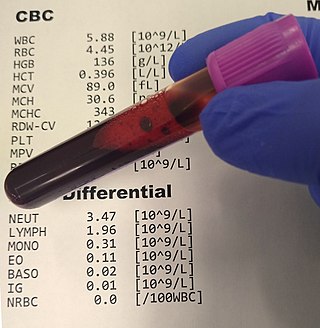
A complete blood count (CBC), also known as a full blood count (FBC), is a set of medical laboratory tests that provide information about the cells in a person's blood. The CBC indicates the counts of white blood cells, red blood cells and platelets, the concentration of hemoglobin, and the hematocrit. The red blood cell indices, which indicate the average size and hemoglobin content of red blood cells, are also reported, and a white blood cell differential, which counts the different types of white blood cells, may be included.

Flow cytometry (FC) is a technique used to detect and measure physical and chemical characteristics of a population of cells or particles.

In biochemistry, immunostaining is any use of an antibody-based method to detect a specific protein in a sample. The term "immunostaining" was originally used to refer to the immunohistochemical staining of tissue sections, as first described by Albert Coons in 1941. However, immunostaining now encompasses a broad range of techniques used in histology, cell biology, and molecular biology that use antibody-based staining methods.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.

A Coulter counter is an apparatus for counting and sizing particles suspended in electrolytes. The Coulter counter is the commercial term for the technique known as resistive pulse sensing or electrical zone sensing. The apparatus is based on the Coulter principle named after its inventor, Wallace H. Coulter.
Fluorescence correlation spectroscopy (FCS) is a statistical analysis, via time correlation, of stationary fluctuations of the fluorescence intensity. Its theoretical underpinning originated from L. Onsager's regression hypothesis. The analysis provides kinetic parameters of the physical processes underlying the fluctuations. One of the interesting applications of this is an analysis of the concentration fluctuations of fluorescent particles (molecules) in solution. In this application, the fluorescence emitted from a very tiny space in solution containing a small number of fluorescent particles (molecules) is observed. The fluorescence intensity is fluctuating due to Brownian motion of the particles. In other words, the number of the particles in the sub-space defined by the optical system is randomly changing around the average number. The analysis gives the average number of fluorescent particles and average diffusion time, when the particle is passing through the space. Eventually, both the concentration and size of the particle (molecule) are determined. Both parameters are important in biochemical research, biophysics, and chemistry.

Acridine orange is an organic compound that serves as a nucleic acid-selective fluorescent dye with cationic properties useful for cell cycle determination. Acridine orange is cell-permeable, which allows the dye to interact with DNA by intercalation, or RNA via electrostatic attractions. When bound to DNA, acridine orange is very similar spectrally to an organic compound known as fluorescein. Acridine orange and fluorescein have a maximum excitation at 502nm and 525 nm (green). When acridine orange associates with RNA, the fluorescent dye experiences a maximum excitation shift from 525 nm (green) to 460 nm (blue). The shift in maximum excitation also produces a maximum emission of 650 nm (red). Acridine orange is able to withstand low pH environments, allowing the fluorescent dye to penetrate acidic organelles such as lysosomes and phagolysosomes that are membrane-bound organelles essential for acid hydrolysis or for producing products of phagocytosis of apoptotic cells. Acridine orange is used in epifluorescence microscopy and flow cytometry. The ability to penetrate the cell membranes of acidic organelles and cationic properties of acridine orange allows the dye to differentiate between various types of cells. The shift in maximum excitation and emission wavelengths provides a foundation to predict the wavelength at which the cells will stain.

Vertico spatially modulated illumination (Vertico-SMI) is the fastest light microscope for the 3D analysis of complete cells in the nanometer range. It is based on two technologies developed in 1996, SMI and SPDM. The effective optical resolution of this optical nanoscope has reached the vicinity of 5 nm in 2D and 40 nm in 3D, greatly surpassing the λ/2 resolution limit applying to standard microscopy using transmission or reflection of natural light according to the Abbe resolution limit That limit had been determined by Ernst Abbe in 1873 and governs the achievable resolution limit of microscopes using conventional techniques.
Cell sorting is the process through which a particular cell type is separated from others contained in a sample on the basis of its physical or biological properties, such as size, morphological parameters, viability and both extracellular and intracellular protein expression. The homogeneous cell population obtained after sorting can be used for a variety of applications including research, diagnosis, and therapy.
Microfluorimetry is an adaption of fluorimetry for studying the biochemical and biophysical properties of cells by using microscopy to image cell components tagged with fluorescent molecules. It is a type of microphotometry that gives a quantitative measure of the qualitative nature of fluorescent measurement and therefore, allows for definitive results that would have been previously indiscernible to the naked eye.
Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit, which is due to the diffraction of light. Super-resolution imaging techniques rely on the near-field or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution or detector-based pixel reassignment, the 4Pi microscope, and structured-illumination microscopy technologies such as SIM and SMI.
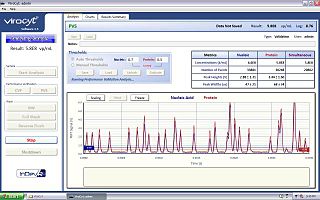
The virus counter is an instrument for rapid quantification of viruses in liquid samples. It is a specialized flow cytometer that uses high-sensitivity fluorescence detection to give a direct measurement of the concentration of virus particles in a fraction of the time required for traditional plaque assays.
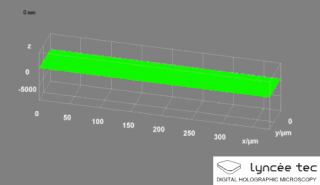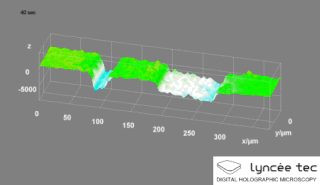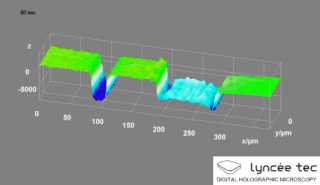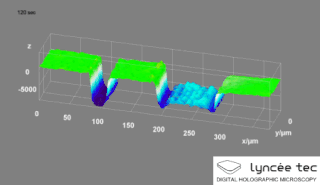
Digital holographic microscopy (DHM) is digital holography applied to microscopy. Digital holographic microscopy distinguishes itself from other microscopy methods by not recording the projected image of the object. Instead, the light wave front information originating from the object is digitally recorded as a hologram, from which a computer calculates the object image by using a numerical reconstruction algorithm. The image forming lens in traditional microscopy is thus replaced by a computer algorithm. Other closely related microscopy methods to digital holographic microscopy are interferometric microscopy, optical coherence tomography and diffraction phase microscopy. Common to all methods is the use of a reference wave front to obtain amplitude (intensity) and phase information. The information is recorded on a digital image sensor or by a photodetector from which an image of the object is created (reconstructed) by a computer. In traditional microscopy, which do not use a reference wave front, only intensity information is recorded and essential information about the object is lost.
Cell counting is any of various methods for the counting or similar quantification of cells in the life sciences, including medical diagnosis and treatment. It is an important subset of cytometry, with applications in research and clinical practice. For example, the complete blood count can help a physician to determine why a patient feels unwell and what to do to help. Cell counts within liquid media are usually expressed as a number of cells per unit of volume, thus expressing a concentration.
Photo-activated localization microscopy and stochastic optical reconstruction microscopy (STORM) are widefield fluorescence microscopy imaging methods that allow obtaining images with a resolution beyond the diffraction limit. The methods were proposed in 2006 in the wake of a general emergence of optical super-resolution microscopy methods, and were featured as Methods of the Year for 2008 by the Nature Methods journal. The development of PALM as a targeted biophysical imaging method was largely prompted by the discovery of new species and the engineering of mutants of fluorescent proteins displaying a controllable photochromism, such as photo-activatible GFP. However, the concomitant development of STORM, sharing the same fundamental principle, originally made use of paired cyanine dyes. One molecule of the pair, when excited near its absorption maximum, serves to reactivate the other molecule to the fluorescent state.

Light sheet fluorescence microscopy (LSFM) is a fluorescence microscopy technique with an intermediate-to-high optical resolution, but good optical sectioning capabilities and high speed. In contrast to epifluorescence microscopy only a thin slice of the sample is illuminated perpendicularly to the direction of observation. For illumination, a laser light-sheet is used, i.e. a laser beam which is focused only in one direction. A second method uses a circular beam scanned in one direction to create the lightsheet. As only the actually observed section is illuminated, this method reduces the photodamage and stress induced on a living sample. Also the good optical sectioning capability reduces the background signal and thus creates images with higher contrast, comparable to confocal microscopy. Because light sheet fluorescence microscopy scans samples by using a plane of light instead of a point, it can acquire images at speeds 100 to 1,000 times faster than those offered by point-scanning methods.
Holotomography (HT) is a laser technique to measure the three-dimensional refractive index (RI) tomogram of a microscopic sample such as biological cells and tissues. Because the RI can serve as an intrinsic imaging contrast for transparent or phase objects, measurements of RI tomograms can provide label-free quantitative imaging of microscopic phase objects. In order to measure 3-D RI tomogram of samples, HT employs the principle of holographic imaging and inverse scattering. Typically, multiple 2D holographic images of a sample are measured at various illumination angles, employing the principle of interferometric imaging. Then, a 3D RI tomogram of the sample is reconstructed from these multiple 2D holographic images by inversely solving light scattering in the sample.
Bacterioplankton counting is the estimation of the abundance of bacterioplankton in a specific body of water, which is useful information to marine microbiologists. Various counting methodologies have been developed over the years to determine the number present in the water being observed. Methods used for counting bacterioplankton include epifluorescence microscopy, flow cytometry, measures of productivity through frequency of dividing cells (FDC), thymidine incorporation, and leucine incorporation.

A white blood cell differential is a medical laboratory test that provides information about the types and amounts of white blood cells in a person's blood. The test, which is usually ordered as part of a complete blood count (CBC), measures the amounts of the five normal white blood cell types – neutrophils, lymphocytes, monocytes, eosinophils and basophils – as well as abnormal cell types if they are present. These results are reported as percentages and absolute values, and compared against reference ranges to determine whether the values are normal, low, or high. Changes in the amounts of white blood cells can aid in the diagnosis of many health conditions, including viral, bacterial, and parasitic infections and blood disorders such as leukemia.


