





DNA damage-binding protein 2 is a protein that in humans is encoded by the DDB2 gene. [5] [6]
DNA damage-binding protein 2 is a protein that in humans is encoded by the DDB2 gene. [5] [6]
As indicated by Rapić-Otrin et al. in 2003, [7] the DDB2 gene is located on human chromosome 11p11.2, spans a region of approximately 24 – 26 kb and includes 10 exons. The DDB2 protein contains five putative WD40 repeats (sequences of about 40 amino acids that can interact with each other) positioned downstream from the second exon. The WD40 motif identified in DDB2 is characteristic of proteins involved in the recognition of chromatin proteins. The C-terminal region of DDB2 (a 48 kDa molecular weight protein) is essential for binding to DDB1 (a larger 127 kDa protein). Together, the two proteins form a UV-damaged DNA binding protein complex (UV-DDB). [8]
If humans have a mutation in each copy of their DDB2 gene, this causes a mild form of the human disease xeroderma pigmentosum, called XPE. [7] Patients in the XPE group have mild dermatological manifestations and are neurologically unaffected. Mutation in the DDB2 gene causes a deficiency in nucleotide excision repair of DNA. This deficiency is also mild, showing 40 to 60% of normal repair capability and a modest sensitivity to UV light in comparison to the sensitivities of cells defective in the other XP genes XPA, XPB, XPC, XPD, XPF and XPG. [9]
As shown by Wittschieben et al., [10] when DDB2 is in a complex with DDB1, forming the heterodimer DDB, this complex binds strongly to DNA containing one type of UV light-induced photoproduct [the (6-4) photoproduct], to DNA with an abasic site, to DNA containing mismatches without a covalent lesion, and to “compound” lesions containing both mismatches and lesions. The heterodimer DDB binds with intermediate strength to DNA containing another UV light-induced photoproduct (the cyclobutane pyrimidine dimer), and binds weakly to DNA that has no DNA damage. The DDB2 component of the heterodimer contains the specificity for binding to damaged DNA, since a heterodimer DDB complex containing amino acid substitutions in the DDB2 subunit, as found in XP-E patients, is very deficient in binding to damaged DNA. DDB1 and DDB2, each acting alone, do not bind DNA.
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow the critical cellular process of DNA repair, the chromatin must be relaxed.
DDB2, in its heterodimeric complex with DDB1, and further complexed with the ubiquitin ligase protein CUL4A [11] and with PARP1 [12] rapidly associates with UV-induced damage within chromatin, with half-maximum association completed in 40 seconds. [11] The PARP1 protein, attached to both DDB1 and DDB2, then PARylates (creates a poly-ADP ribose chain) on DDB2 that attracts the DNA remodeling protein ALC1. [12] Action of ALC1 relaxes the chromatin at the site of UV damage to DNA. This relaxation allows other proteins in the nucleotide excision repair pathway to enter the chromatin and repair the DNA damaged by the UV-induced presence of cyclobutane pyrimidine dimers.
In 2015, Zhu et al. [13] showed that DDB2 down-regulates the acetylation of lysine 56 in histone H3 (H3K56Ac) after UV-induced DNA damage through DDB2 interaction with histone deacetylases 1 and 2. Decreased acetylation of histones decreases transcription of associated genes in the DNA wrapped around the histones.
In 2016, Zou et al. [14] showed that DDB2 is involved in cell cycle arrest and homologous recombinational DNA repair after cells are subjected to ionizing radiation.
In 2016, Christmann et al. [15] showed that exposure of cells to the carcinogenic benzo(a)pyrene metabolite BPDE caused prompt and sustained upregulation of DDB2. This contributed to enhanced removal of BPDE adducts from DNA.
In 2017, Fantini et al. [16] showed that DDB2, in association with XRCC5 and XRCC6 (otherwise known as Ku80 and Ku70, which make up the Ku heterodimer), has transcriptional activities. The DDB2/Ku effects on transcription are separate from the actions of the Ku heterodimer in non-homologous end joining DNA repair.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.
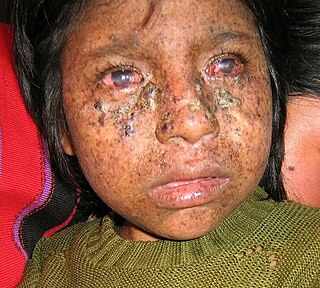
Xeroderma pigmentosum (XP) is a genetic disorder in which there is a decreased ability to repair DNA damage such as that caused by ultraviolet (UV) light. Symptoms may include a severe sunburn after only a few minutes in the sun, freckling in sun-exposed areas, dry skin and changes in skin pigmentation. Nervous system problems, such as hearing loss, poor coordination, loss of intellectual function and seizures, may also occur. Complications include a high risk of skin cancer, with about half having skin cancer by age 10 without preventative efforts, and cataracts. There may be a higher risk of other cancers such as brain cancers.

Nucleotide excision repair is a DNA repair mechanism. DNA damage occurs constantly because of chemicals, radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.

XPB is an ATP-dependent DNA helicase in humans that is a part of the TFIIH transcription factor complex.
The family of heterochromatin protein 1 (HP1) consists of highly conserved proteins, which have important functions in the cell nucleus. These functions include gene repression by heterochromatin formation, transcriptional activation, regulation of binding of cohesion complexes to centromeres, sequestration of genes to the nuclear periphery, transcriptional arrest, maintenance of heterochromatin integrity, gene repression at the single nucleosome level, gene repression by heterochromatization of euchromatin, and DNA repair. HP1 proteins are fundamental units of heterochromatin packaging that are enriched at the centromeres and telomeres of nearly all eukaryotic chromosomes with the notable exception of budding yeast, in which a yeast-specific silencing complex of SIR proteins serve a similar function. Members of the HP1 family are characterized by an N-terminal chromodomain and a C-terminal chromoshadow domain, separated by a hinge region. HP1 is also found at some euchromatic sites, where its binding can correlate with either gene repression or gene activation. HP1 was originally discovered by Tharappel C James and Sarah Elgin in 1986 as a factor in the phenomenon known as position effect variegation in Drosophila melanogaster.

TFIIH subunit XPD is a protein that in humans is encoded by the ERCC2 gene. It is a component of the general transcription and DNA repair factor IIH (TFIIH) core complex involved in transcription-coupled nucleotide excision repair.

DNA excision repair protein ERCC-1 is a protein that in humans is encoded by the ERCC1 gene. Together with ERCC4, ERCC1 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

Cullin-4A is a protein that in humans is encoded by the CUL4A gene. CUL4A belongs to the cullin family of ubiquitin ligase proteins and is highly homologous to the CUL4B protein. CUL4A regulates numerous key processes such as DNA repair, chromatin remodeling, spermatogenesis, haematopoiesis and the mitotic cell cycle. As a result, CUL4A has been implicated in several cancers and the pathogenesis of certain viruses including HIV. A component of a CUL4A complex, Cereblon, was discovered to be a major target of the teratogenic agent thalidomide.

UV excision repair protein RAD23 homolog B is a protein that in humans is encoded by the RAD23B gene.

Xeroderma pigmentosum, complementation group C, also known as XPC, is a protein which in humans is encoded by the XPC gene. XPC is involved in the recognition of bulky DNA adducts in nucleotide excision repair. It is located on chromosome 3.

DNA repair protein complementing XP-A cells is a protein that in humans is encoded by the XPA gene.

DNA excision repair protein ERCC-6 is a protein that in humans is encoded by the ERCC6 gene. The ERCC6 gene is located on the long arm of chromosome 10 at position 11.23.

DNA repair protein complementing XP-G cells is a protein that in humans is encoded by the ERCC5 gene.

DNA damage-binding protein 1 is a protein that in humans is encoded by the DDB1 gene.

Cullin-4B is a protein that in humans is encoded by the CUL4B gene which is located on the X chromosome. CUL4B has high sequence similarity with CUL4A, with which it shares certain E3 ubiquitin ligase functions. CUL4B is largely expressed in the nucleus and regulates several key functions including: cell cycle progression, chromatin remodeling and neurological and placental development in mice. In humans, CUL4B has been implicated in X-linked intellectual disability and is frequently mutated in pancreatic adenocarcinomas and a small percentage of various lung cancers. Viruses such as HIV can also co-opt CUL4B-based complexes to promote viral pathogenesis. CUL4B complexes containing Cereblon are also targeted by the teratogenic drug thalidomide.

DNA excision repair protein ERCC-8 is a protein that in humans is encoded by the ERCC8 gene.

Transcription initiation protein SPT3 homolog is a protein that in humans is encoded by the SUPT3H gene.

DNA polymerase eta, is a protein that in humans is encoded by the POLH gene.
DNA damage-binding protein or UV-DDB is a protein complex that is responsible for repair of UV-damaged DNA. This complex is composed of two protein subunits, a large subunit DDB1 (p127) and a small subunit DDB2 (p48). When cells are exposed to UV radiation, DDB1 moves from the cytosol to the nucleus and binds to DDB2, thus forming the UV-DDB complex. This complex formation is highly favorable and it is demonstrated by UV-DDB's binding preference and high affinity to the UV lesions in the DNA. This complex functions in nucleotide excision repair, recognising UV-induced (6-4) pyrimidine-pyrimidone photoproducts and cyclobutane pyrimidine dimers.
DNA damage is an alteration in the chemical structure of DNA, such as a break in a strand of DNA, a nucleobase missing from the backbone of DNA, or a chemically changed base such as 8-OHdG. DNA damage can occur naturally or via environmental factors, but is distinctly different from mutation, although both are types of error in DNA. DNA damage is an abnormal chemical structure in DNA, while a mutation is a change in the sequence of base pairs. DNA damages cause changes in the structure of the genetic material and prevents the replication mechanism from functioning and performing properly. The DNA damage response (DDR) is a complex signal transduction pathway which recognizes when DNA is damaged and initiates the cellular response to the damage.