Related Research Articles

Crystallography is the experimental science of determining the arrangement of atoms in crystalline solids. Crystallography is a fundamental subject in the fields of materials science and solid-state physics. The word "crystallography" is derived from the Greek words crystallon "cold drop, frozen drop", with its meaning extending to all solids with some degree of transparency, and graphein "to write". In July 2012, the United Nations recognised the importance of the science of crystallography by proclaiming that 2014 would be the International Year of Crystallography.

X-ray crystallography is the experimental science determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract into many specific directions. By measuring the angles and intensities of these diffracted beams, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal. From this electron density, the mean positions of the atoms in the crystal can be determined, as well as their chemical bonds, their crystallographic disorder, and various other information.
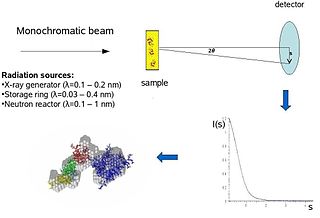
Biological small-angle scattering is a small-angle scattering method for structure analysis of biological materials. Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers. Small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) are the two complementary techniques known jointly as small-angle scattering (SAS). SAS is an analogous method to X-ray and neutron diffraction, wide angle X-ray scattering, as well as to static light scattering. In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles. When used to study biological materials, which are very often in aqueous solution, the scattering pattern is orientation averaged.

Jerome Karle was an American physical chemist. Jointly with Herbert A. Hauptman, he was awarded the Nobel Prize in Chemistry in 1985, for the direct analysis of crystal structures using X-ray scattering techniques.
In physics, the phase problem is the problem of loss of information concerning the phase that can occur when making a physical measurement. The name comes from the field of X-ray crystallography, where the phase problem has to be solved for the determination of a structure from diffraction data. The phase problem is also met in the fields of imaging and signal processing. Various approaches of phase retrieval have been developed over the years.
Electron crystallography is a method to determine the arrangement of atoms in solids using a transmission electron microscope (TEM).
Multi-wavelength anomalous diffraction is a technique used in X-ray crystallography that facilitates the determination of the three-dimensional structure of biological macromolecules via solution of the phase problem.
Acta Crystallographica is a series of peer-reviewed scientific journals, with articles centred on crystallography, published by the International Union of Crystallography (IUCr). Originally established in 1948 as a single journal called Acta Crystallographica, there are now six independent Acta Crystallographica titles:

In molecular biology, a RING (short for Really Interesting New Gene) finger domain is a protein structural domain of zinc finger type which contains a C3HC4 amino acid motif which binds two zinc cations (seven cysteines and one histidine arranged non-consecutively). This protein domain contains 40 to 60 amino acids. Many proteins containing a RING finger play a key role in the ubiquitination pathway.
Multiple isomorphous replacement (MIR) is historically the most common approach to solving the phase problem in X-ray crystallography studies of proteins. For protein crystals this method is conducted by soaking the crystal of a sample to be analyzed with a heavy atom solution or co-crystallization with the heavy atom. The addition of the heavy atom to the structure should not affect the crystal formation or unit cell dimensions in comparison to its native form, hence, they should be isomorphic.
Resolution in terms of electron density is a measure of the resolvability in the electron density map of a molecule. In X-ray crystallography, resolution is the highest resolvable peak in the diffraction pattern, while resolution in cryo-electron microscopy is a frequency space comparison of two halves of the data, which strives to correlate with the X-ray definition.
Single-wavelength anomalous diffraction (SAD) is a technique used in X-ray crystallography that facilitates the determination of the structure of proteins or other biological macromolecules by allowing the solution of the phase problem. In contrast to multi-wavelength anomalous diffraction, SAD uses a single dataset at a single appropriate wavelength. One advantage of the technique is the minimization of time spent in the beam by the crystal, thus reducing potential radiation damage to the molecule while collecting data. SAD is sometimes called "single-wavelength anomalous dispersion", but no dispersive differences are used in this technique since the data are collected at a single wavelength. Today, selenium-SAD is commonly used for experimental phasing due to the development of methods for selenomethionine incorporation into recombinant proteins.

Protein crystallization is the process of formation of a regular array of individual protein molecules stabilized by crystal contacts. If the crystal is sufficiently ordered, it will diffract. Some proteins naturally form crystalline arrays, like aquaporin in the lens of the eye.

Crystallographic image processing (CIP) is traditionally understood as being a set of key steps in the determination of the atomic structure of crystalline matter from high-resolution electron microscopy (HREM) images obtained in a transmission electron microscope (TEM) that is run in the parallel illumination mode. The term was created in the research group of Sven Hovmöller at Stockholm University during the early 1980s and became rapidly a label for the "3D crystal structure from 2D transmission/projection images" approach. Since the late 1990s, analogous and complementary image processing techniques that are directed towards the achieving of goals with are either complementary or entirely beyond the scope of the original inception of CIP have been developed independently by members of the computational symmetry/geometry, scanning transmission electron microscopy, scanning probe microscopy communities, and applied crystallography communities.
In molecular biology, S4 domain refers to a small RNA-binding protein domain found in a ribosomal protein named uS4. The S4 domain is approximately 60-65 amino acid residues long, occurs in a single copy at various positions in different proteins and was originally found in pseudouridine syntheses, a bacterial ribosome-associated protein.
The term macromolecular assembly (MA) refers to massive chemical structures such as viruses and non-biologic nanoparticles, cellular organelles and membranes and ribosomes, etc. that are complex mixtures of polypeptide, polynucleotide, polysaccharide or other polymeric macromolecules. They are generally of more than one of these types, and the mixtures are defined spatially, and with regard to their underlying chemical composition and structure. Macromolecules are found in living and nonliving things, and are composed of many hundreds or thousands of atoms held together by covalent bonds; they are often characterized by repeating units. Assemblies of these can likewise be biologic or non-biologic, though the MA term is more commonly applied in biology, and the term supramolecular assembly is more often applied in non-biologic contexts. MAs of macromolecules are held in their defined forms by non-covalent intermolecular interactions, and can be in either non-repeating structures, or in repeating linear, circular, spiral, or other patterns. The process by which MAs are formed has been termed molecular self-assembly, a term especially applied in non-biologic contexts. A wide variety of physical/biophysical, chemical/biochemical, and computational methods exist for the study of MA; given the scale of MAs, efforts to elaborate their composition and structure and discern mechanisms underlying their functions are at the forefront of modern structure science.
Ribosomal L28e protein family is a family of evolutionarily related proteins. Members include 60S ribosomal protein L28.
In crystallography, direct methods is a set of techniques used for structure determination using diffraction data and a priori information. It is a solution to the crystallographic phase problem, where phase information is lost during a diffraction measurement. Direct methods provides a method of estimating the phase information by establishing statistical relationships between the recorded amplitude information and phases of strong reflections.
Quantum crystallography is a branch of crystallography that investigates crystalline materials within the framework of quantum mechanics, with analysis and representation, in position or in momentum space, of quantities like wave function, electron charge and spin density, density matrices and all properties related to them. Like the quantum chemistry, Quantum crystallography involves both experimental and computational work. The theoretical part of quantum crystallography is based on quantum mechanical calculations of atomic/molecular/crystal wave functions, density matrices or density models, used to simulate the electronic structure of a crystalline material. While in quantum chemistry, the experimental works mainly rely on spectroscopy, in quantum crystallography the scattering techniques play the central role, although spectroscopy as well as atomic microscopy are also sources of information.
This is a timeline of crystallography.
References
- 1 2 "The Nobel Prize in Chemistry 1985". NobelPrize.org. Retrieved 2020-03-20.
- ↑ Usón I, Sheldrick GM (1999). "Advances in direct methods for protein crystallography". Curr. Opin. Struct. Biol. 9 (5): 643–8. doi: 10.1016/S0959-440X(99)00020-2 . PMID 10508770.
- ↑ Hauptman H (1997). "Phasing methods for protein crystallography". Curr. Opin. Struct. Biol. 7 (5): 672–80. doi:10.1016/S0959-440X(97)80077-2. PMID 9345626.
- ↑ "(IUCr) Crystallographic software list". www.iucr.org. Retrieved 2020-03-20.
| | This crystallography-related article is a stub. You can help Wikipedia by expanding it. |