Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.
Anovulation is when the ovaries do not release an oocyte during a menstrual cycle. Therefore, ovulation does not take place. However, a woman who does not ovulate at each menstrual cycle is not necessarily going through menopause. Chronic anovulation is a common cause of infertility.
Hypogonadism means diminished functional activity of the gonads—the testicles or the ovaries—that may result in diminished production of sex hormones. Low androgen levels are referred to as hypoandrogenism and low estrogen as hypoestrogenism. These are responsible for the observed signs and symptoms in both males and females.
Kallmann syndrome (KS) is a genetic disorder that prevents a person from starting or fully completing puberty. Kallmann syndrome is a form of a group of conditions termed hypogonadotropic hypogonadism. To distinguish it from other forms of hypogonadotropic hypogonadism, Kallmann syndrome has the additional symptom of a total lack of sense of smell (anosmia) or a reduced sense of smell. If left untreated, people will have poorly defined secondary sexual characteristics, show signs of hypogonadism, almost invariably are infertile and are at increased risk of developing osteoporosis. A range of other physical symptoms affecting the face, hands and skeletal system can also occur.
Isolated hypogonadotropic hypogonadism (IHH), also called idiopathic or congenital hypogonadotropic hypogonadism (CHH), as well as isolated or congenital gonadotropin-releasing hormone deficiency (IGD), is a condition which results in a small subset of cases of hypogonadotropic hypogonadism (HH) due to deficiency in or insensitivity to gonadotropin-releasing hormone (GnRH) where the function and anatomy of the anterior pituitary is otherwise normal and secondary causes of HH are not present.
X-linked adrenal hypoplasia congenita is a genetic disorder that mainly affects males. It involves many endocrine tissues in the body, especially the adrenal glands.
Chromosome instability syndromes are a group of inherited conditions associated with chromosomal instability and breakage. They often lead to an increased tendency to develop certain types of malignancies.
The gonadotropin-releasing hormone receptor (GnRHR), also known as the luteinizing hormone releasing hormone receptor (LHRHR), is a member of the seven-transmembrane, G-protein coupled receptor (GPCR) family. It is the receptor of gonadotropin-releasing hormone (GnRH). The GnRHR is expressed on the surface of pituitary gonadotrope cells as well as lymphocytes, breast, ovary, and prostate.
Anosmin-1 is a secreted, EM associated glycoprotein found in humans and other organisms responsible for normal development, which is expressed in the brain, spinal cord and kidney. Absence or damage to the protein results in Kallmann syndrome in humans, which is characterized by loss of olfactory bulbs and GnRH secretion leading to anosmia and hypothalamic hypogonadotropic hypogonadism. Anosmin-1 is coded by the KAL-1 gene, which is found on the X chromosome. Anosmin-1 is 100 kilodaltons and is expressed on the outside of cells. Because of this and because of its contribution to normal migration of nerve cells, a role in the extracellular matrix has been postulated.

DAX1 is a nuclear receptor protein that in humans is encoded by the NR0B1 gene. The NR0B1 gene is located on the short (p) arm of the X chromosome between bands Xp21.3 and Xp21.2, from base pair 30,082,120 to base pair 30,087,136.

Gonadotropin-releasing hormone receptor is a protein that in humans is encoded by the GNRHR gene.

Follitropin subunit beta also known as follicle-stimulating hormone beta subunit (FSH-B) is a protein that in humans is encoded by the FSHB gene. Alternative splicing results in two transcript variants encoding the same protein.
Progonadoliberin-2 is a protein that in humans is encoded by the GNRH2 gene.

Thyroid stimulating hormone, beta also known as TSHB is a protein which in humans is encoded by the TSHB gene.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.
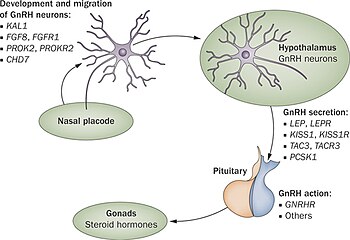
Hypogonadotropic hypogonadism (HH), is due to problems with either the hypothalamus or pituitary gland affecting the hypothalamic-pituitary-gonadal axis. Hypothalamic disorders result from a deficiency in the release of gonadotropic releasing hormone (GnRH), while pituitary gland disorders are due to a deficiency in the release of gonadotropins from the anterior pituitary. GnRH is the central regulator in reproductive function and sexual development via the HPG axis. GnRH is released by GnRH neurons, which are hypothalamic neuroendocrine cells, into the hypophyseal portal system acting on gonadotrophs in the anterior pituitary. The release of gonadotropins, LH and FSH, act on the gonads for the development and maintenance of proper adult reproductive physiology. LH acts on Leydig cells in the male testes and theca cells in the female. FSH acts on Sertoli cells in the male and follicular cells in the female. Combined this causes the secretion of gonadal sex steroids and the initiation of folliculogenesis and spermatogenesis. The production of sex steroids forms a negative feedback loop acting on both the anterior pituitary and hypothalamus causing a pulsatile secretion of GnRH. GnRH neurons lack sex steroid receptors and mediators such as kisspeptin stimulate GnRH neurons for pulsatile secretion of GnRH.

GnRH neurons, or gonadotropin-releasing hormone expressing neurons, are the cells in the brain that control the release of reproductive hormones from the pituitary. These brain cells control reproduction by secreting GnRH into the hypophyseal portal capillary bloodstream, so are sometimes referred to as “sex neurons”. This small capillary network carries GnRH to the anterior pituitary, causing release of luteinizing hormone (LH) and follicle stimulating hormone (FSH) into the wider bloodstream. When GnRH neurons change their pattern of release from the juvenile to the adult pattern of GnRH secretion, puberty is initiated. Failure of GnRH neurons to form the proper connections, or failure to successfully stimulate the pituitary with GnRH, means that puberty is not initiated. These disruptions to the GnRH system cause reproductive disorders like hypogonadotropic hypogonadism or Kallmann Syndrome.

FEZ family zinc finger 1 is a protein that in humans is encoded by the FEZF1 gene.

The fertile eunuch syndrome or Pasqualini syndrome is a cause of hypogonadotropic hypogonadism caused by a luteinizing hormone deficiency. It is characterized by hypogonadism with spermatogenesis. Pasqualini and Bur published the first case of eunuchoidism with preserved spermatogenesis in 1950 in la Revista de la Asociación Médica Argentina. The hypoandrogenism with spermatogenesis syndrome included:

Boucher-Neuhäuser syndrome is a very rare genetic disorder which is characterized by a triad consisting of cerebellar ataxia, chorioretinal dystrophy, and hypogonadism.