
Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in the Mediterranean fever (MEFV) gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.

Caspase-1/Interleukin-1 converting enzyme (ICE) is an evolutionarily conserved enzyme that proteolytically cleaves other proteins, such as the precursors of the inflammatory cytokines interleukin 1β and interleukin 18 as well as the pyroptosis inducer Gasdermin D, into active mature peptides. It plays a central role in cell immunity as an inflammatory response initiator. Once activated through formation of an inflammasome complex, it initiates a proinflammatory response through the cleavage and thus activation of the two inflammatory cytokines, interleukin 1β (IL-1β) and interleukin 18 (IL-18) as well as pyroptosis, a programmed lytic cell death pathway, through cleavage of Gasdermin D. The two inflammatory cytokines activated by Caspase-1 are excreted from the cell to further induce the inflammatory response in neighboring cells.

Muckle–Wells syndrome (MWS) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives, and can lead to amyloidosis. Individuals with MWS often have episodic fever, chills, and joint pain. As a result, MWS is considered a type of periodic fever syndrome. MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin. MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease—in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS).
Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.

NLR family pyrin domain containing 3 (NLRP3), is a protein that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.

Etiocholanolone, also known as 5β-androsterone, as well as 3α-hydroxy-5β-androstan-17-one or etiocholan-3α-ol-17-one, is an etiocholane (5β-androstane) steroid as well as an endogenous 17-ketosteroid that is produced from the metabolism of testosterone. It causes fever, immunostimulation, and leukocytosis, and is used to evaluate adrenal cortex function, bone marrow performance, and in neoplastic disease to stimulate the immune system. Etiocholanolone is also known to be an inhibitory androstane neurosteroid, acting as a positive allosteric modulator of the GABAA receptor, and possesses anticonvulsant effects. The unnatural enantiomer of etiocholanolone is more potent as a positive allosteric modulator of GABAA receptors and as an anticonvulsant than the natural form.

Mevalonate kinase deficiency (MKD) is an autosomal recessive metabolic disorder that disrupts the biosynthesis of cholesterol and isoprenoids. It is a rare genetic disorder, but a high frequency is observed in Northern European regions.

NRAS is an enzyme that in humans is encoded by the NRAS gene. It was discovered by a small team of researchers led by Robin Weiss at the Institute of Cancer Research in London. It was the third RAS gene to be discovered, and was named NRAS, for its initial identification in human neuroblastoma cells.
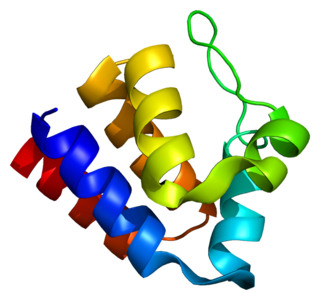
PYCARD, often referred to as ASC, is a protein that in humans is encoded by the PYCARD gene. It is localized mainly in the nucleus of monocytes and macrophages. In case of pathogen infection, however, it relocalizes rapidly to the cytoplasm, perinuclear space, endoplasmic reticulum and mitochondria and it is a key adaptor protein in activation of the inflammasome.

NLRP1 encodes NACHT, LRR, FIIND, CARD domain and PYD domains-containing protein 1 in humans. NLRP1 was the first protein shown to form an inflammasome. NLRP1 is expressed by a variety of cell types, which are predominantly epithelial or hematopoietic. The expression is also seen within glandular epithelial structures including the lining of the small intestine, stomach, airway epithelia and in hairless or glabrous skin. NLRP1 polymorphisms are associated with skin extra-intestinal manifestations in CD. Its highest expression was detected in human skin, in psoriasis and in vitiligo. Polymorphisms of NLRP1 were found in lupus erythematosus and diabetes type 1. Variants of mouse NLRP1 were found to be activated upon N-terminal cleavage by the protease in anthrax lethal factor.

NLR family CARD domain-containing protein 4 is a protein that in humans is encoded by the NLRC4 gene.

NACHT, LRR and PYD domains-containing protein 2 is a protein that in humans is encoded by the NLRP2 gene.
NACHT, LRR and PYD domains-containing protein 7 is a protein that in humans is encoded by the NLRP7 gene.
Nucleotide-binding oligomerization domain-like receptor (NLR) pyrin domain (PYD)-containing protein 12 is a protein that in humans is encoded by the NLRP12 gene.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.
A pyrin domain is a protein domain and a subclass of protein motif known as the death fold, the 4th and most recently discovered member of the death domain superfamily (DDF). It was originally discovered in the pyrin protein, or marenostrin, encoded by MEFV. The mutation of the MEFV gene is the cause of the disease known as Familial Mediterranean Fever. The domain is encoded in 23 human proteins and at least 31 mouse genes.

NLRP10, short for NOD-like receptor family pyrin domain containing 10, is an intracellular protein of mammals that functions in apoptosis and the immune system. It is also known as NALP10, NOD8, PAN5, Pynod, and CLR11.1, and is one of 14 pyrin domain containing members of the NOD-like receptor family of cytoplasmic receptors, although it differs from other NOD-like receptors by lacking the characteristic leucine-rich repeat domain. It is also believed that it helps regulate the inflammatory response. NLRP10 reduces inflammatory and innate immune responses by inhibiting the activity of two proteins associated with the inflammasome; caspase-1 and PYCARD.
NOD-like receptor family pyrin domain containing 11 is a protein that in humans is encoded by the NLRP11 gene located on the long arm of human chromosome 19q13.42. NLRP11 belongs to the NALP subfamily, part of a large subfamily of CATERPILLER. It is also known as NALP11, PYPAF6, NOD17, PAN10, and CLR19.6
NLRP (Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing), also abbreviated as NALP, is a type of NOD-like receptor. NOD-like receptors are a type of pattern recognition receptor that are found in the cytosol of the cell, recognizing signals of antigens in the cell. NLRP proteins are part of the innate immune system and detect conserved pathogen characteristics, or pathogen-associated molecular patterns, such as such as peptidoglycan, which is found on some bacterial cells. It is thought that NLRP proteins sense danger signals linked to microbial products, initiating the processes associated with the activation of the inflammasome, including K+ efflux and caspase 1 activation. NLRPs are also known to be associated with a number of diseases. Research suggests NLRP proteins may be involved in combating retroviruses in gametes. As of now, there are at least 14 different known NLRP genes in humans, which are named NLRP1 through NLRP14. The genes translate into proteins with differing lengths of leucine-rich repeat domains.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by dysfunction of the innate immune system. These responses are characterized by periodic or chronic systemic inflammation, usually without the involvement of adaptive immunity.




